Abstract
Genetically modified models have been proven to be a powerful tool in drug discovery. The ability to genetically modify the mouse genome by removing or replacing a specific gene has enhanced our ability to identify and validate target genes of interest. Humanized mouse models include genetic modifications at the gene level ranging from single base changes to complete replacement of the murine gene with an orthologous human gene. Humanization can also occur at the cell and organ level, i.e. bone marrow transplantation models. The need for more predictive in vivo models to evaluate the efficacy, pharmacokinetic profile, and toxological properties of lead compounds has made humanized mice an emerging critical tool. In addition, the recent focus on development of species specific biotherapeutic molecules has resulted in the recognition of humanized mice as essential in the drug discovery process.
Introduction
Among animal species, the laboratory mouse has been instrumental in modeling human biology and disease. It is well-documented that the mouse is a powerful tool in the understanding of human biological pathways such as hematopoiesis as well as providing insights into human diseases, such as oncogenesis, neurological disorders and cardiovascular and metabolic diseases [1]. In addition, over the last twenty years, technological advances have facilitated the genetic engineering of the mouse genome (Table 1). Examples of these genetic modifications include deletion of a gene from the mouse genome (knockout), replacement of a murine gene with its human ortholog (knock-in), and addition of a unique human gene to the murine genome (transgenic). More recently, the ability to populate the mouse liver with human hepatocytes (humanized liver) and to engraft mouse bone marrow with human hematopoietic stem cells (humanized bone marrow) has also provided exciting new opportunities [2,3]. In this article, we discuss the use of humanized mouse models in drug discovery and how they have led to a better understanding of disease pathways and ultimately improved the translational value of preclinical studies.
Understanding Disease Pathways and Designing Therapeutic Interventions Using Mouse Models
Historically, the oncology field relied heavily on xenograft models to test cancer therapeutics. However, xenograft models are inherently limited, as the cells are from cancer cell lines that have been maintained in a culture dish and may differ from cancers in situ in a patient. In addition, in a xenograft model, the tumor stroma of vasculature, extracellular matrix and immune cell infiltration is supplied by the host mouse and may not truly reflect the interplay between oncogenic cells and the stroma in a human environment. Owing to these limitations, xenograft models are thought to be partly responsible for the exceptionally high phase II attrition rate seen in oncology trials [4]. However, in recent years, a new generation of mouse models has been generated with the goal to reproduce human oncogenic processes. These models are making a positive impact in elucidating disease etiology, tumor progression and metastasis.

Acute promyelocytic leukemia (APL) serves as an example of a disease where the use of mouse models facilitated the development of an effective targeted therapy for this once fatal disease. APL is a cancer of the bone marrow and blood and is characterized by an accumulation of the immature granulocytes called promyelocytes. At the genome level [5], APL carries the somatic mutation of a chromosomal translocation between the retinoic acid receptor alpha (RARα) gene located on q11.2 of chromosome 17 (human gene ID: 6256) and among 95% of APL cases, the promyelocytic leukemia (PML) gene residing on q22 of chromosome 15 (human gene ID:5371), t(15;17) (q22;q11.2). As a result of this reciprocal translocation, two fusion genes are created, the PML-RARα and RARα-PML. To establish the causal role of each of the fusion genes in APL, several groups resorted to the use of transgenic mouse technology. In these experiments, the PML-RARα fusion cDNA was placed under the control of the human cathespin G (hCG) [6,7] or the migration inhibitory factor-related protein 8 (MRP8) promoters [8] which then direct PML-RARα expression to the myeloid lineage of cells. Mice expressing the PML-RARα transgene have altered myeloid development characterized by increased percentages of immature over mature myeloid cells in the blood, bone marrow, and spleen. After a latent period of 6 months or longer, approximately 10% to 30% of the mice developed acute myeloid leukemia.
It was demonstrated that, similar to patients carrying the PML-RARα translocation, the PML-RARα transgenic mice respond to all-trans retinoic acid (ATRA) therapy, suggesting that the PML-RARα mice could be considered as a surrogate model in which to test therapeutics for human APL [9]. Using the PML-RARα transgenic mice, it was discovered that a combination treatment of ATRA with arsenic trioxide (As2O3) was very eff ective compared to either alone. It was also noticed that similar to patients carrying another fusion, PLZF-RARα the PLZF-RARα transgenic mice responded poorly to ATRA. Later studies showed that histone deacetylase inhibitors, such as phenylbutyrate, in combination with ATRA, induced remission of APL in these transgenic mice [10]. These drug combinations may therefore be effective in APL patients carrying the PLZF-RARα translocation.
A third aspect of the APL mouse model use is that it has also been instrumental in identifying additional mutations that drive APL oncogenesis. It was noticed early on that the PML-RARα transgenic mice develop APL after a latent period of 6 months to more than a year and with 10% to 30% penetrance, suggesting the need for additional mutations to progress APL. Human tumor cells typically carry hundreds of alterations at the genome level, with the majority acquired throughout the course of a patient’s life. It could be challenging to screen through these alterations and identify the mutation(s) that cooperate with PML-RARα to drive APL. In contrast, a transgenic mouse model could be created on an inbred background, making it a simpler task to screen through the list of diff erences between the cancer and the normal reference genomes, for which sequences of several inbred strains of mice, such as that of 129 and C57BL/6, are publically accessible [11].
Historically, quantitative trait locus (QTL) analysis and position cloning had been used to identify a region or a locus in the genome responsible for or contributing to a phenotypic trait. With the advancement in genome sequencing technology, it is now routinely possible to sequence an entire genome. By sequencing the APL genomes of a PML-RARα knockin mouse model, several somatic, nonsynonymous mutations likely relevant to APL pathogenesis were discovered [12], including a Jak1 V657F (V658F in human) mutation and a 150-kb deletion involving the lysine (K)-specific demethylase 6A (Kdm6a) gene. Identification of these additional genes and validation of their roles in driving APL should lead to a better understanding of APL disease pathways and most importantly, to identification of additional drug targets that may offer further therapeutic opportunities for treating APL.
Efficacy and PK/PD Studies Enabled by Humanized Mouse Models
The successful development of compounds for the treatment of thrombocytopenia has relied heavily on the availability of humanized mouse models. Thrombocytopenia refers to a condition in which blood platelet count is below 50,000 per microliter, as compared to the 150,000 to 450,000 counts per microliter found among the normal human population. It is a phenotype associated with medical disorders that suppress or decrease thrombopoiesis or accelerate platelet destruction, such as immune suppression, autoimmunity and severe liver diseases. Severity of the phenotype can range from bruising to intracranial hemorrhage.
Thrombopoietin is a cytokine produced in the liver and exerts its effect by binding to the thrombopoietin receptor (TPOR, C-MPL, CD110) (human gene ID:1234) expressed on CD34+ hematopoietic progenitors and the megakaryocytic lineage cells to regulate thrombopoiesis [13,14]. TPOR, thus, presents an attractive target against which to develop therapeutics for the treatment of thrombocytopenia. To this end, orally active, nonpeptidyl agonists of TPOR have been reported by several groups, including those from Pfizer [15-18]. The first TPOR agonist, Eltrombopag (SB-497115-GR) of GlaxoSmithKline [15], was approved for the treatment of idiopathic thrombocytopenic purpura (ITP) in 2008.
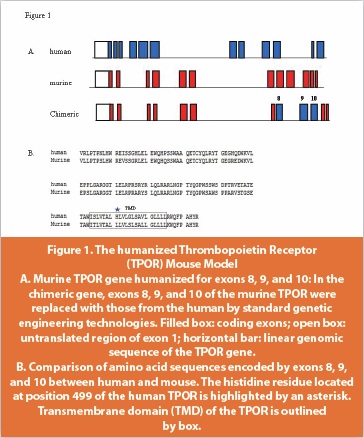
In preclinical studies, it was discovered that these orally active, nonpeptidyl TPOR agonists only bind to the human and chimpanzee TPOR, making it impossible to assess their efficacy and toxicity using conventional preclinical animal models including rodents and monkeys. Further studies suggested that a single histidine residue located within the transmembrane domain of the human and chimpanzee TPOR (histidine 499) is responsible for this specificity and this histidine residue is not present in laboratory animal species, including mice, guinea pigs and cynomolgus monkeys [18].
Although preclinical studies can be performed in chimpanzees [19], this species is not easily accessible and experimental use is costly. Therefore, other systems were investigated. In one study, researchers used immunodeficient mouse models engrafted with human hematopoietic stem cells [16]. The NOG (NOD/Shi-scid/IL-2Rγnull) host mice are on the NOD (non obese diabetic) background and also carry two other mutations, the scid and IL-2Rγ knockout. As a result, they are severely immunodeficient and can thus support engraftment of xenogenic donor bone marrow to a high degree. To generate these mice, the host bone marrow is first obliterated by γ-irradiation and then these mice are injected with the donor CD34+ human hematopoietic stem cells (HSCs). In the host mice, the donor HSCs engraft and undergo hematopoiesis and give rise to cells of multilineages including the megakaryocytic lineage. The authors reported successful maintenance of approximately 3% human platelets in the peripheral circulation and 50% of human megakaryocytes in bone marrow of these engrafted mice for up to 1 year. This humanized bone marrow mouse model was used to study efficacy and PK/PD properties of TPOR agonists [16,20].
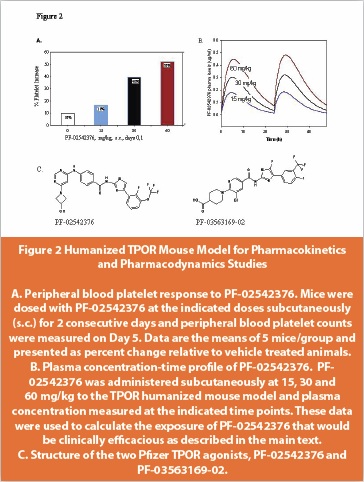
We reported the discovery of a series of pyrimidine benzamide-based compounds that exhibit TPOR agonist properties [17,21]. Similar to the findings from others, our compounds also exhibit human TPOR binding specificity. To develop an animal model for our program, we created a mouse model carrying a chimeric TPOR gene in which the leucine codon at position 490 of the murine gene would be replaced with the histidine codon at position 499 of the human gene (Figure 1). When administered to our humanized TPOR mouse model, the Pfizer lead TPOR agonist, PF-02542376 [17], was shown to increase platelet counts in the TPOR humanized mice but not the wild type animals and the effect seen was dose dependent (Figure 2).
In addition, the TPOR humanized mouse model was used for another compound PF-03563169-02 [21], to obtain an initial estimate of the concentration that would be required for efficacy in the clinic. In this case, the PF-03563169-02 AUC (area under curve) that was associated with a 50% increase in platelet counts was approximately 10 μg*h/ml, equivalent to a 24-hour average concentration of 0.42 μg/ml (0.77 μM). Based on these parameters, a clinically efficacious exposure of PF-03563169-02 was estimated to be 0.77 μM (0.42 μg/ml).
Bone Marrow Transplantation Models for Gene Therapy
As mentioned above, host mice that are severely immunodeficient (NSG or NOD) can be engrafted with peripheral blood mononuclear cells (PBMC) or human CD34+ HSCs [2]. The fact that these mice carry human CD4+ helper T cells makes them particularly useful for HIV research and drug discovery. Human immunodeficiency virus 1 (HIV) infection leads to the acquired immunodeficiency syndrome (AIDS) which is a major health issue around the globe. It is known that the HIV-1 virus gains entry into CD4+ helper T cells by binding to the CD4 receptor and the CCR5 chemokine coreceptor (human gene ID:4352). The discovery that people homozygous for the mutation CCR5Δ32 are resistant to HIV-1 infection [22] suggests that blocking or deleting CCR5 could be an effective and safe therapeutic strategy. Maraviroc (UK-427857), an “entry inhibitor” developed by Pfizer, is an allosteric modulator of the CCR5 coreceptor [23]. This drug was approved by the FDA in 2007 for the treatment of AIDS.
Deletion of CCR5 has also been actively pursued as a potential gene therapy strategy. Zinc finger nucleases (ZFN) are chimeric proteins created through genetic engineering and consist of a zinc finger domain which confers DNA binding specificity and the Fok-1 restriction endonuclease domain which creates a double strand break in DNA [24]. In this scheme, a pair of ZFN targeting the CCR5 locus was designed and shown to cut the CCR5 gene in primary human CD4+ cells [25] or primary human CD34+ hematopoietic stem cells [26]. After electroporation of the ZFN pair targeting CCR5, the human cells were transplanted and allowed to engraft the host mice. Subsequently, mice were infected with HIV-1. Data showed that cells retaining an intact CCR5 gene were selectively eliminated, while those in which the CCR5 gene had been deleted were spared and left to proliferate and populate the host mice. Thus, one can envision a gene therapy strategy for HIV patients – treating a patient’s CD4+ helper T cells or CD34+ HSC cells with a pair of ZFN targeting the CCR5 gene and transplantation of these cells back into the patient. Recently, a clinical stage biopharmaceutical company announced positive phase I results regarding its CCR5-modified, autologous, T-cell product (SB-728-T) for the treatment of HIV/AIDS. In developing this therapeutic strategy, the mouse model with the humanized bone marrow played an instrumental role in testing potential therapeutics.
Other Model Species
In addition to mice, other genetically modified model species are making their way into the laboratory. Recently, knockout of the cystic fibrosis transmembrane conductance regulator (CFTR) (human gene ID: 1080) gene in the pig has been reported [27], resulting in a pig model of human cystic fibrosis. This was accomplished by disrupting the CFTR gene in fibroblasts of outbred domestic pigs by homologous recombination and then transferring the genetically modified nucleus into an enucleated pig oocyte (somatic cell nuclear transfer) to generate CFTR knockout pigs. The pig is closer in size to humans and may be a preferable model for cardiovascular and metabolic diseases.
The rat is also a preferred preclinical model for studying drug metabolism, drug safety, CNS disorders, diabetes, inflammation and oncology [28]. Regrettably, for the past two decades, the use of genetically modified rats in drug discovery has been severely hampered, due to the lack of a germline competent rat embryonic stem cell line. It was not until recently that germline competent rat embryonic stem cell lines have been isolated and shown to be amenable to homologous recombination, as suggested by the successful deletion of the tumor suppressor p53 gene [29]. To add to the tool box, targeted mutagenesis in rat using zinc finger nucleases [30], and transcription activator-like effector nuclease technologies [31] which circumvent the need of a germline competent embryonic stem cell line, has been achieved. It is conceivable that, in the near future, more sophisticated genetic engineering in the rats will be accomplished and rat models will be generated to support drug discovery to the same degree that mice have over the last 20 years.
Conclusion
Mice are the most widely used animal species in drug discovery. When used appropriately, the mouse can be a powerful in-vivo system in which to dissect disease pathways as well as improve our understanding of pharmacological interventions as exemplified by the cases discussed above.
However, the translational value of the mouse model largely depends on whether the disease pathway under investigation is conserved between the two species. Thus, the more we understand these pathways, the better we can assess whether the mouse is the correct and best preclinical species for the study of interest. This point is made all too well by the tragedy of the “London Trial” in 2006 [32], when all six healthy human subjects developed an unexpected catastrophic cytokine storm and went into multiple organ failure shortly after being administered the compound TGN1412, a humanized monoclonal antibody of the IgG4 subclass directed against the human CD28 antigen. This reaction in humans was unexpected, as studies in all preclinical animal models, including mice and cynomolgus monkeys, had shown that the compound was safe. Since then, many hypotheses have been offered to explain these different responses between humans and the preclinical species used. Recent data [33] suggest that, although the CD28 receptor from cynomolgus monkey shares identical amino acid sequences in the extracellular domain and binds TGN1412 with similar affinity as that in the human, it is not expressed in the effector memory T cell population, the suspected cell source for cytokine storm in humans.
Thus, the choice of a preclinical species suitable for the disease pathway under investigation should be carefully weighed against several considerations. Not only the ortholog relationship between the two species for the target gene of interest, but also the cell type expressing the gene, the level of gene expression, and ultimately, conservation of the biological function at the organ and system level must be carefully considered.
Acknowledgement
We wish to thank William H. Brissette and Sharon L. Ripp for providing PK/ PD data using the TPOR knock in mouse model.
References
1. Zambrowicz, B.P. and Sands, A.T. (2003) Knockouts model the 100 best-selling drugs--will they model the next 100? Nat Rev Drug Discov 2 (1), 38-51
2. Shultz, L.D. et al. (2007) Humanized mice in translational biomedical research. Nat Rev Immunol 7 (2), 118-130
3. Melkus, M.W. et al. (2006) Humanized mice mount specifi c adaptive and innate immune responses to EBV and TSST-1. Nat Med 12 (11), 1316-1322
4. Kola, I. and Landis, J. (2004) Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov 3 (8), 711-715
5. Kakizuka, A. et al. (1991) Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RAR alpha with a novel putative transcription factor, PML. Cell 66 (4), 663-674
6. Grisolano, J.L. et al. (1997) Altered myeloid development and acute leukemia in transgenic mice expressing PML-RAR alpha under control of cathepsin G regulatory sequences. Blood 89 (2), 376-387
7. He, L.Z. et al. (1997) Acute leukemia with promyelocytic features in PML/RARalpha transgenic mice. Proc Natl Acad Sci U S A 94 (10), 5302-5307
8. Brown, D. et al. (1997) A PMLRARalpha transgene initiates murine acute promyelocytic leukemia. Proc Natl Acad Sci U S A 94 (6), 2551-2556
9. Rego, E.M. et al. (2000) Retinoic acid (RA) and As2O3 treatment in transgenic models of acute promyelocytic leukemia (APL) unravel the distinct nature of the leukemogenic process induced by the PML-RARalpha and PLZF-RARalpha oncoproteins. Proc Natl Acad Sci U S A 97 (18), 10173-10178
10. He, L.Z. et al. (2001) Histone deacetylase inhibitors induce remission in transgenic models of therapyresistant acute promyelocytic leukemia. J Clin Invest 108 (9), 1321-1330
11. Waterston, R.H. et al. (2002) Initial sequencing and comparative analysis of the mouse genome. Nature 420 (6915), 520-562
12. Wartman, L.D. et al. (2011) Sequencing a mouse acute promyelocytic leukemia genome reveals genetic events relevant for disease progression. J Clin Invest 121 (4), 1445-1455
13. Vigon, I. et al. (1992) Molecular cloning and characterization of MPL, the human homolog of the v-mpl oncogene: identification of a member of the hematopoietic growth factor receptor superfamily. Proc Natl Acad Sci U S A 89 (12), 5640-5644
14. Mignotte, V. et al. (1994) Structure and transcription of the human c-mpl gene (MPL). Genomics 20 (1), 5-12
15. Erickson-Miller, C.L. et al. (2005) Discovery and characterization of a selective, nonpeptidyl thrombopoietin receptor agonist. Exp Hematol 33 (1), 85-93
16. Nakamura, T. et al. (2006) A novel nonpeptidyl human c-Mpl activator stimulates human megakaryopoiesis and thrombopoiesis. Blood 107 (11), 4300-4307
17. Reiter, L.A. et al. (2007) Pyrimidine benzamide-based thrombopoietin receptor agonists. Bioorg Med Chem Lett 17 (19), 5447-5454
18. Yamane, N. et al. (2008) Characterization of novel non-peptide thrombopoietin mimetics, their species specificity and the activation mechanism of the thrombopoietin receptor. Eur J Pharmacol 586 (1-3), 44-51
19. Erickson-Miller, C.L. et al. (2009) Preclinical activity of eltrombopag (SB-497115), an oral, nonpeptide thrombopoietin receptor agonist. Stem Cells 27 (2), 424-430
20. Nogami, W. et al. (2008) The effect of a novel, small non-peptidyl molecule butyzamide on human thrombopoietin receptor and megakaryopoiesis. Haematologica 93 (10), 1495-1504
21. Antipas, A.S. et al. (2010) Structure-activity relationships and hepatic safety risks of thiazole agonists of the thrombopoietin receptor. Bioorg Med Chem Lett 20 (14), 4069-4072
22. Liu, R. et al. (1996) Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 86 (3), 367-377
23. Dorr, P. et al. (2005) Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob Agents Chemother 49 (11), 4721-4732
24. Kim, Y.G. et al. (1996) Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci U S A 93 (3), 1156-1160
25. Perez, E.E. et al. (2008) Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol 26 (7), 808-816
26. Holt, N. et al. (2010) Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol 28 (8), 839-847
27. Rogers, C.S. et al. (2008) Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science 321 (5897), 1837-1841
28. Aitman, T.J. et al. (2008) Progress and prospects in rat genetics: a community view. Nat Genet 40 (5), 516-522
29. Tong, C. et al. (2010) Production of p53 gene knockout rats by homologous recombination in embryonic stem cells. Nature 467 (7312), 211-213
30. Geurts, A.M. et al. (2009) Knockout rats via embryo microinjection of zinc-finger nucleases. Science 325 (5939), 433
31. Tesson, L. et al. (2011) Knockout rats generated by embryo microinjection of TALENs. Nat Biotechnol 29 (8), 695-696
32. Suntharalingam, G. et al. (2006) Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med 355 (10), 1018-1028
33. Eastwood, D. et al. (2010) Monoclonal antibody TGN1412 trial failure explained by species differences in CD28 expression on CD4+ effector memory T-cells. Br J Pharmacol 161 (3), 512-526
34. Smithies, O. et al. (1985) Insertion of DNA sequences into the human chromosomal beta-globin locus by homologous recombination. Nature 317 (6034), 230-234
35. Thomas, K.R. et al. (1986) High frequency targeting of genes to specific sites in the mammalian genome. Cell 44 (3), 419-428
36. Kuehn, M.R. et al. (1987) A potential animal model for Lesch-Nyhan syndrome through introduction of HPRT mutations into mice. Nature 326 (6110), 295-298
37. Brinster, R.L. and Palmiter, R.D. (1984) Transgenic mice containing growth hormone fusion genes. Philos Trans R Soc Lond B Biol Sci 307 (1132), 309-312
38. Traggiai, E. et al. (2004) Development of a human adaptive immune system in cord blood celltransplanted mice. Science 304 (5667), 104-107
39. Shultz, L.D. et al. (2005) Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol 174 (10), 6477-6489
Author Biographies
Dr. Rosalba E. Sacca received her Ph.D. in Biology from the Albert Einstein College of Medicine in New York and did postdoctoral fellowships at the Massachusetts Institute of Technology in Cambridge, MA and the Memorial Sloan-Kettering Cancer Center in New York. Prior to joining Pfizer Dr. Sacca was an Associate Research Scientist at Yale University where she researched the role of cytokines in autoimmune disease. Dr. Sacca joined Pfizer in 1998 as a Research Scientist in the Genetically Modified Models group and in 2004 assumed the role of Associate Director with responsibilities for overseeing the efforts and managing the global portfolio of the GeMM group. She also served as chair of the steering committees for several strategic external collaborations. In 2006 she became Director in the Biology Discipline in Discovery Research where her responsibilities included support of the Biology CoEs business strategies and strategic imperatives. In 2007 Rosalba joined the Worldwide Exploratory Science & Technology group as Director of Scientific Analysis and Coordination, where she served as scientific liaison and alliance manager for several key academic alliances, including collaborations with Scripps and UCSF. Dr. Sacca in 2008 took on the role of Director and head of the Genetically Modified Models Center of Excellence (GeMM CoE) group in Worldwide Research & Development at Pfizer, Inc. Her responsibilities included development and application of all genetically modified animal and stem cell technologies for Pfizer’s global R&D to support target validation and disease model development for drug discovery. Dr. Sacca currently works as Director of Scientific Development for a global provider of research products and services.
Dr. Wenning Qin holds the position of Associate Director for a laboratory that specializes in mammalian genetics research. Dr. Qin is responsible for developing genetically engineered animal models. She has over 10 years of experience designing and creating genetically engineered animal models supporting drug discovery and development, including experiences with the Monsanto Company, Pharmacia Corporation and Pfizer Inc. She holds a Ph.D. in Pharmacology from Southern Illinois University (Carbondale, IL.).
Following graduation from the Purdue University School of Science, Jeffrey Stock joined the laboratory of Dr. S. Steven Potter at the Cincinnati Children’s Hospital Medical Center and focused his work on the development of genetically modified mouse models to study murine development. In 1992, Jeff joined the newly created Genetically Modified Models group at Pfizer Inc. During the 20 years with Pfizer, Jeff has developed genetically modified mouse models supporting all therapeutic groups within Pfizer’s global R&D. Jeff has also contributed to over 20 publications characterizing the genetically modified models he has developed during his 25 years in the field.