Introduction
Amorphous pharmaceuticals represent both opportunity and necessity in pharmaceutical development. The opportunity arises from the potential to improve bioavailability via use of an amorphous form, rather than a crystalline form. On the other hand, it is sometimes the case that no crystalline form is available, in which case it is necessary to deal with the amorphous form. This article will discuss the development of amorphous pharmaceuticals, with reference to three case studies from within AstraZeneca.
The use of high throughput methods in drug discovery has led to compounds with more lipophilic properties and hence poor aqueous solubility [1,2], resulting in drugs with dissolution-limited bioavailability. In the case of poorly soluble but well-permeable drugs (BCS class II), high free energy states such as the amorphous form can significantly improve ‘apparent’ solubility [3,4], often leading to large increases in dissolution rate in the gastro-intestinal tract, thus increasing the bioavailability. However, since the amorphous form is a highly metastable state, there is a thermodynamic drive towards crystallization; in some cases even at temperatures below the glass transition temperature (Tg) [5]. In cases where a crystalline drug has been rendered amorphous, it is common practice to prepare a dispersion of the drug in a pharmaceutically-acceptable polymer in order to stabilize against crystallization [6]. In some cases, the use of a dispersion may have a further role in acting as a crystallization inhibitor in vivo [7].
It is sometimes the case that no crystalline form, including salts, co-crystals or pharmaceutically-acceptable solvates, is available. The amorphous drug may be isolated by a variety of methods including precipitation or desolvation of a solvate, and the ease of isolation will be affected by the glass transition temperature (Tg) and the extent to which the Tg is lowered by residual solvent [8]. In some cases, amorphous salts have been found to have a higher Tg than the free acid or base [9,10], and amorphous dispersions may also be used to improve the physical properties.
A brief overview of the preparation and characterization of amorphous pharmaceuticals will be followed by case studies of (A) an amorphous drug requiring no stabilization, (B) a poorly-soluble drug (BCS class II) in which an amorphous dispersion is used to achieve improved bioavailability and maintain physical stability and (C) an amorphous drug for which no crystalline forms are known.
Preparation and Characterization of Amorphous Pharmaceuticals
Amorphous materials can be made in a variety of ways including mechanical treatment, heating (quench cooling or desolvation) and various solvent-based processes (including rotary evaporation, lyophilization, precipitation and spray-drying). The case studies here all used spray-drying to produce amorphous API or solid dispersions in quantities ranging from a few grams to tens of kilograms [11]. Spray-drying has the benefit of exposing the sample to a high temperature for only a very short time and of providing samples in powdered form requiring no particle size reduction. On the other hand, spray-drying requires some process development to achieve high yields, and it can sometimes be difficult to find a solvent suitable for both the drug and the polymer(s) of interest. In assessing the quality of spray-dried material, an initial, qualitative assessment will typically be based on examination by Polarized Light Microscopy (PLM), Powder X-Ray Diffraction (XRPD) and Differential Scanning Calorimetry (DSC). These analytical techniques can rule out materials which clearly contain crystalline drug. Further development of an amorphous form may require quantification of the level of crystallinity, in particular to determine the level of detection and set a specification for manufacture (described here for Compound B).
XRPD and vibrational spectroscopy have been used to quantify the amount of crystalline in amorphous drug [12-15], with levels of detection as low as 0.5 and 1%, depending on the system investigated and the method used, and detection limits of 0.2% have been achieved by XRPD with synchrotron radiation [16]. Solid State Nuclear Magnetic Resonance (ssNMR) has also been used to analyze crystalline and amorphous content [17-19].
For both XRPD and vibrational spectroscopy it is necessary to prepare a calibration set by mixing crystalline API with a suitable amorphous matrix, and this itself is not a trivial exercise. Some of the difficulties which may be encountered include: ensuring that the crystalline standard is representative of the crystalline material present in the samples, achieving homogeneity of standards and the physical stability of the standards. ssNMR has the advantage of being a standardless method; the amount of crystalline material is given by the relative areas of the sharp peaks due to the crystalline content and the broad peaks of the amorphous component.
Understanding whether an amorphous drug or dispersion is stable with respect to crystallization in the long term is difficult to address. Storage under stressed conditions such as 40oC/75%RH is often used in order to quickly rule out drugs or dispersions that are unstable. However, it is recognized that these conditions may not relate to the long-term storage conditions, particularly since a sample may be taken above the Tg by a combination of moisture sorption and elevated temperature. In the case of dispersions, phase separation may indicate a lack of long- term stability, and this may be indicated by the presence of more than one Tg in DSC measurements (for instance, that of the pure drug as well as that of the dispersion).
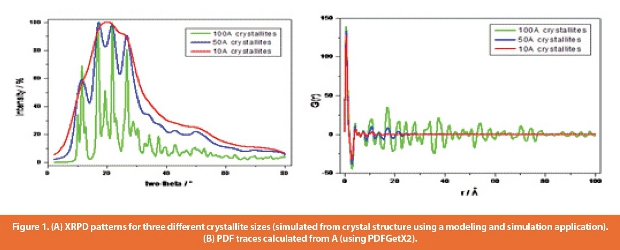
A recent addition to the tools available for characterization of amorphous drugs and dispersions is the pair-wise distribution function approach (PDF). This takes into account all the scattered radiation, including the “halo” observed for X-ray amorphous samples [20] and describes the probability of finding two atoms separated by a certain distance, r. The method provides information on short-range order as well as long-range, which makes it of particular value in understanding amorphous materials. Historically the PDF method has been applied to inorganic materials using synchrotron X-Ray sources, but recently interest has focused on the application to pharmaceutical materials using laboratory X-Ray sources [21,22].
Figure 1 compares the PDF traces of crystalline, amorphous and nanocrystalline forms of a compound; the XRPD’s were simulated from the crystal structure using a modeling and simulation application and then the PDF traces were calculated using PDFGetX2 [23]. It can be seen that for the amorphous example, the PDF indicates the length range over which the sample is ordered; in this case 10A, while the powder pattern simply shows that the sample is amorphous.
Case studies
Compound A: An Amorphous API Not Requiring Stabilization
Compound A is an amorphous HCl salt, prepared by spray-drying a crystalline solvate of the salt. Following small-scale lab trials, the compound was spray-dried at the 50g scale, using methanol as the solvent and achieving a yield of 83%. Secondary drying was carried out and this achieved a residual methanol content of between 0.05% and 0.8%, depending on batch and scale of manufacture.
 The dry Tg of the compound was 127oC (measured after in-situ drying in the DSC). However, the presence of residual solvent lowered the Tg to values between 68 and 80oC; the exact value varied from batch to batch depending on the amount of residual solvent.
The dry Tg of the compound was 127oC (measured after in-situ drying in the DSC). However, the presence of residual solvent lowered the Tg to values between 68 and 80oC; the exact value varied from batch to batch depending on the amount of residual solvent.
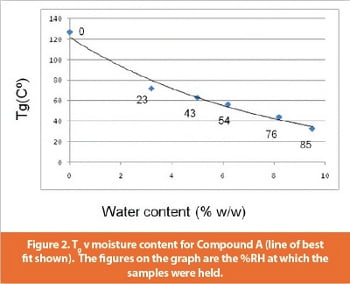
An investigation was carried out into the effect of relative humidity and moisture sorption on the Tg and handling properties. This was achieved by storing samples over saturated salt solutions for several days and then measuring moisture content and Tg; results are shown in Figure 2.
This study demonstrated that the drug should be stored with dessicant, and confirmed that secondary drying should aim to reduce the solvent content as low as practicable in order to maintain the Tg as high as possible. Given the degree of the plasticization effect of moisture on the compound, the question of whether the compound could be handled under ambient conditions and still remain a freeflowing powder was addressed with further testing. A sample was held at 25oC/60%RH for 17 days, (at which point the Tg was measured as 52oC); microscopy showed that the morphology of the particles had not changed and the sample remained a powder, giving an indication that it would be possible to handle the compound under ambient conditions.
The spray-dried material was of high purity and chemical stability testing demonstrated that storage at room temperature with desiccant would give an adequate shelf-life. In this case, the amorphous compound is relatively straightforward to develop for several reasons: the immediate precursor is crystalline (which helps to provide high purity), the isolation route is a straightforward desolvation by spraydrying, and the Tg of the material is sufficiently high that problems in handling are avoided.
Compound B: Improving the Bioavailability of a Poorly-soluble Crystalline Drug
The second study, compound B, is a crystalline drug in BCS Class II, i.e. bioavailability limited by dissolution; at doses higher than 50mg, bioavailability is variable and non-linear, and so use of amorphous drug was investigated. The amorphous drug had a Tg of 54oC, (prepared by quench-cooling in the DSC and measured under dry conditions), and crystallization was seen to commence, at room temperature, very soon after amorphous preparation. It was therefore clearly necessary to stabilize the amorphous drug against crystallization.
A series of dispersions of Compound B with various polymers was prepared at drug loadings of 20 to 50% drug w/w. These were initially examined by XRPD and DSC, to rule out dispersions which were clearly not effective; for instance, those with a relatively high drug loading were seen to crystallize on heating during DSC measurement, or gave crystalline XRPD peaks after a short storage time.  The remaining dispersions were evaluated by stability testing, in which samples were stored at a range of conditions including forced degradation conditions. Samples were tested for physical stability by XRPD and DSC, and dissolution testing was carried out to confirm that amorphous dispersions did confer advantages over the crystalline form. The lead dispersion that emerged from this work was a dispersion with HPMCP (hydroxypropyl methyl cellulose phthalate) with a drug loading of 25% w/w.
The remaining dispersions were evaluated by stability testing, in which samples were stored at a range of conditions including forced degradation conditions. Samples were tested for physical stability by XRPD and DSC, and dissolution testing was carried out to confirm that amorphous dispersions did confer advantages over the crystalline form. The lead dispersion that emerged from this work was a dispersion with HPMCP (hydroxypropyl methyl cellulose phthalate) with a drug loading of 25% w/w.
The dispersion was manufactured by spray-drying from a solution of acetone containing approximately 12% solids (drug and polymer combined). Secondary drying was necessary to achieve acceptable solvent levels, and roller compaction was used to densify the dispersion for formulation. Figure 3 shows the morphology of the dispersion before and after roller compaction.
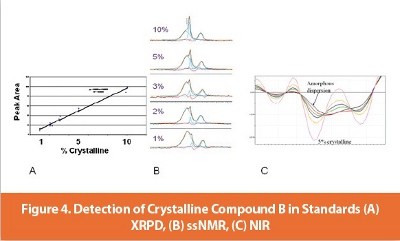 Quantitative methods for the detection of crystalline drug in dispersion were developed for the spray-dried powder. A set of standards containing 1, 2, 3, 5 and 10% crystalline drug by weight was prepared. Using this set of standards, it was possible to devise an XRPD method with a level of detection of 1% crystalline drug. A NIR method was also developed; this method was in good agreement with XRPD, had a slightly lower limit of detection than XRPD and has the potential to determine water and acetone content at the same time as crystallinity. 19F ssNMR was also run on these standards, and it was demonstrated that 1% crystallinity could be detected. Note that the limit of detection of 1% crystalline is relative to the dispersion and not the drug; since the drug is 25% of the dispersion, then the level of detection, expressed as a fraction of the drug, is 4%.
Quantitative methods for the detection of crystalline drug in dispersion were developed for the spray-dried powder. A set of standards containing 1, 2, 3, 5 and 10% crystalline drug by weight was prepared. Using this set of standards, it was possible to devise an XRPD method with a level of detection of 1% crystalline drug. A NIR method was also developed; this method was in good agreement with XRPD, had a slightly lower limit of detection than XRPD and has the potential to determine water and acetone content at the same time as crystallinity. 19F ssNMR was also run on these standards, and it was demonstrated that 1% crystallinity could be detected. Note that the limit of detection of 1% crystalline is relative to the dispersion and not the drug; since the drug is 25% of the dispersion, then the level of detection, expressed as a fraction of the drug, is 4%.
Analysis of stability samples showed that crystallization occurred under forced degradation conditions but not at 25oC /60%RH (up to six months). Dissolution testing showed that dissolution failures correlated with the presence of crystalline material. However, a problem with stability testing was recognized: under forced degradation conditions the polymer was not stable and suffered from impurity growth and a reduction in molecular weight distribution. It is therefore possible that in this case, accelerated stability testing is inappropriate.
Dissolution testing showed that the amorphous dispersion gave higher solution concentrations at high-dose than did formulations of the crystalline drug (Figure 5). The impact on bioavailability was that the exposure achieved by the amorphous dispersion was higher than that from the crystalline drug, and continued to increase as dose increased.
We have retrospectively made simple dispersions of Drug B with various polymers, and applied the PDF analysis method, with promising results. 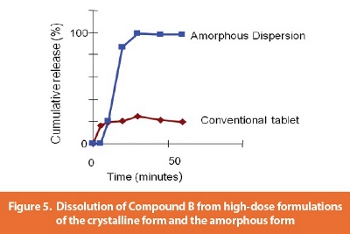 Examination of the freshly-prepared dispersion by standard XRPD shows little distinction between the two dispersions while the PDF suggests that the local order in the second dispersion extends to a slightly longer scale than in the first dispersion. The dispersions were then held at 40oC/75%RH for 15 days. Optical microscopy (imaged under crossed-polars) shows crystallization has occurred in the second dispersion, while the first dispersion has remained largely free from crystallization.
Examination of the freshly-prepared dispersion by standard XRPD shows little distinction between the two dispersions while the PDF suggests that the local order in the second dispersion extends to a slightly longer scale than in the first dispersion. The dispersions were then held at 40oC/75%RH for 15 days. Optical microscopy (imaged under crossed-polars) shows crystallization has occurred in the second dispersion, while the first dispersion has remained largely free from crystallization.
This example of Compound B illustrates that it is possible to stabilize a drug with a low Tg and high propensity to crystallize. However, this requires a rather high carrier to drug ratio, which may be problematic if a high dose is required.
Compound C Case Study: An Unstable Amorphous Compound without a Crystalline Form
In some cases it is necessary to develop an amorphous pharmaceutical because no crystalline forms can be identified. As mentioned above, the ease of development of such a compound will be critically dependant on the Tg, both in the dry state and at typical levels of solvent.  Compound C is an amorphous compound which had been subject to many attempts to find a crystalline form, whether the free base or as a salt, but all attempts were unsuccessful. The drug had a Tg of 70oC when dry but at typical levels of solvent this could drop to 15oC, and this led to difficulties in isolating and handling the amorphous free base.
Compound C is an amorphous compound which had been subject to many attempts to find a crystalline form, whether the free base or as a salt, but all attempts were unsuccessful. The drug had a Tg of 70oC when dry but at typical levels of solvent this could drop to 15oC, and this led to difficulties in isolating and handling the amorphous free base.
Initial precipitation attempts failed to isolate the amorphous solid from the liquors, since the precipitated particles deliquesced on the filter. However, a low temperature precipitation method was developed in which moisture was excluded and the solid maintained at a temperature below the Tg during all stages of precipitation and isolation. In this process, a solution of the drug was added to an anti-solvent at -15°C, during which time particles were formed. The solvents were then distilled off before filtering the slurry under cold nitrogen.
Spray-drying of the drug was successful on the lab-scale, although the low Tg of the compound limited the choice of solvent, but the yield was low and the process likely to be impractical at a manufacturing scale since it required ‘open loop’ conditions, i.e. use of fresh, rather than recirculated N2 gas, in order to dry the material effectively. 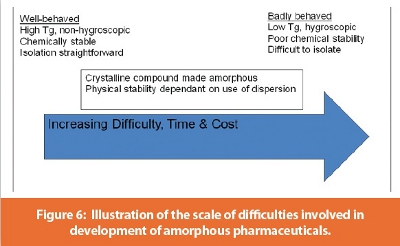
Some success in improving the ease of precipitation and physical properties was achieved by forming amorphous salts. The salts prepared, and their Tg values are given in Table 1. Although these salts generally gave an increase in Tg compared to the free base, they did not achieve adequate chemical stability and so were not progressed further.
Dispersions of the drug with a range of polymers and cyclodextrins (β-CD and HPβCD) were prepared by spray-drying. The polymers used included: HPMCP, PVP, PVA and copovidone, and these were spray-dried at a drug loading of 10%w/w, while the cyclodextrins were spray-dried at stoichiometric ratios. Despite generally improving the physical properties of the compound, most dispersions failed to give an improvement in chemical stability. However, three dispersions were found to combine an improvement in physical properties with acceptable chemical stability; these were PVA and the cyclodextrins.
This case study demonstrates the burden placed on the drug development process by a ‘difficult’ amorphous compound. In the case of Compound C, there is no crystallization process immediately prior to isolation of the amorphous free base to confer purity, the compound has poor chemical stability, low Tg, has an affinity for solvent and is hygroscopic. 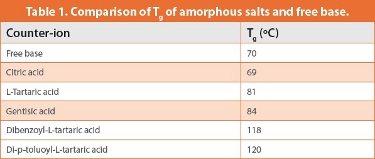 These properties led to difficulties in isolating the AFB itself, even at small scale, ruled out amorphous salts because of their poor chemical stability, and severely restricted the carriers that could be used in spray-drying. This particular compound however, did have one advantage, and that was its low dose, which would allow a low drug loading in a dispersion.
These properties led to difficulties in isolating the AFB itself, even at small scale, ruled out amorphous salts because of their poor chemical stability, and severely restricted the carriers that could be used in spray-drying. This particular compound however, did have one advantage, and that was its low dose, which would allow a low drug loading in a dispersion.
Conclusion
Amorphous pharmaceuticals present the industry with a mixture of opportunities and challenges, ranging from improvements in the bioavailability of poorly soluble drugs to the isolation and formulation of compounds which will not crystallize and have poor physical properties. These three case studies demonstrate the range of difficulties associated with the development of amorphous API with a range of behaviors from ‘well-behaved’ to ‘badly-behaved’.
‘Well-behaved’ compounds with an acceptably high Tg, good chemical stability and straightforward isolation route, such as Compound A should be relatively easy to develop, while at the other extreme, compounds such as C, with low Tg, poor chemical stability and a complex isolation route will present many challenges. Compounds in which a crystalline material is rendered amorphous and possibly stabilized by a carrier (Compound B) represent the intermediate case; the challenge here is clearly to prevent crystallization and ensure that the solubility advantage is maintained and translated into a biopharmaceutical advantage.
Acknowledgements
The authors are grateful to Steve Cosgrove, David Gray, Dedong Wu, Paul Stott, Steve Holland, Alessia Portieri, Liz Meehan, Jan Cherryman, Richard Storey (AstraZeneca) and Sarah Gibson (Cambridge University) for sharing data and helpful discussions.
References
1. G. M. Keseru and G. M. Makara, ‘The Infl uence of Lead discovery Strategies on the Properties of drug Candidates.’ Nature Reviews, Drug Discovery, 8 (2009) 203.
2. C. A. Lipinski, Journal of Pharmacological and Toxicological Methods, 44 (2000), 235
3. B. C. Hancock and M. Parks, ‘What is the true solubility advantage for amorphous pharmaceuticals?’ Pharm Res 17 (2000) 397.
4. B. C. Hancock and G. Zografi , ‘Characteristics and Signifi cance of the Amorphous state’, J. Pharm. Sci., 86, 1997, pp1-12.
5. T. Wu and L. Yu, ‘Surface Crystallization of Indomethacin Below Tg’. Pharm Res 23 (2006) 2350
6. K. Nagapudi and J. Jona, ‘Amorphous Active Pharmaceutical Ingredients in Preclinical Studies: Preparation, Characterization and Formulation’, Current Bioactive Compounds, 4 (2008) 213
7. H. R. Guzman, M. Tawa, Z. Zhang, P. Ratanabanangkoon, P. Shaw, C. Gardner, H. Chen, J Moreau, O. Almarsson and J. F. Remenar, ‘Combined use of crystalline salt forms and precipitation inhibitors to improve oral absorption of celecoxib from solid oral formulations’ J. Pharm. Sci., 96(10) (2007) 2686.
8. B.C. Hancock and G. Zografi , ‘The Relationship Between the Glass Transition Temperature and the Water Content of Amorphous Pharmaceutical Solids’, Pharm. Res. 11(4) (1994) 471
9. C.S.Towler, T. Li., H. Wikstrom, D. M. Remick, M. V. Sanchez and L. S. Taylor, ‘An Investigation into the Properties of Some Amorphous Organic Salts’ Molecular Pharmaceutics, 5 (2008) 946
10. P. Tong, L.S. Taylor and G.Zografi , ‘Influence of Alkali Metal Counterions on the Glass Transition Temperature of Amorphous Indomethacin Salts’, Pharm Res, 2002 (19) 649
11. I. McConvey, M. Hoyle and O. Nulty, 2009, ‘Reducing the hazards associated with spray drying from laboratory through to larger scale including considerations for outsourcing and secondary containmentI IChemE Symposium Series, Hazards XXI
12. L.S. Taylor and G. Zografi , ‘The Quantitative Analysis of Crystallinity Using FT-Raman Spectroscopy’ , Pharm Res, 15(5) (1998) 755
13. R. Surana and R. Suryanarayanan, ‘Quantitation of Crystallinity in Substantially Amorphous Pharmaceuticals and Study of Crystallization Kinetics by X-Ray Powder Diffractometry’, Powder Diffraction 15(1) (2000) 2
14. I. Fix and K-J. Steffens, ‘Quantifying Low amorphous or Crystalline Amounts of Alpha- Lactose-Monohydrarte Using X-Ray Powder Diffraction, Near Infrared Spectroscopy and Differential Scanning Calorimetry’ Drug Dev Ind Pharm, 30(5) (2004) 513
15. C. Clunes, A. Mahendrasingram and R Suryanarayanan, ‘Quantification of Crystallinity in Substantially Amorphous Materials by Synchrotron X-Ray Powder Diffractometry’ Pharm Res, 22(11) (2005) 1942
16. A. Heinz, C.J. Strachan, K.C. Gordon and T. Rades, ‘Analysis of Solid State Transformations of Pharmaceutical Compounds Using Vibrational Spectroscopy’ J. Pharm. Pharmacol, 61 (2009) 971
17. R. Lefort, A. De Gusseme, J-F Wilart, F. Danede, and M. Descamps, ’Solid state NMR and DSC Methods for Quantifying the Amorphous Content in Solid Dosage Forms: an Application to Ball- Milling of Trehalose’ I. J. Pharma 280 (2004) 209.
18. R.K. Harris, ’NMR Studies of Organic Polymorphs and Solvates’, The Analyst, 131 (2006) 351
19. S.J. Byard, S. L. Jackson, A. Smail, M. Bauer, D.C. Apperley, ’Studies on the Crystallinity of a Pharmaceutical Development Drug Substance’ J. Pharm. Sci. 94 (2005) 1321
20. T. Egami and S. J. L. Billinge, Underneath the Bragg Peaks: Structural Analysis of Complex Materials, Elsevier Science, Oxford, 2004.
21. S. Bates, G. Zografi , D. Engers, K. Morris, K. Crowley and A. Newman, Analysis of Amorphous and Nanocrystalline Solids from their X-Ray Diff raction Patterns’ Pharm Res, 23 (2006) 2333
22. A. Newman, D. Engers, S.Bates, I. Ivanisevic, R.C. Kelly and G. Zografi , ‘Characterization of Amorphous API:Polymer Mixtures Using X-Ray Powder Diff raction’, J. Pharm. Sci, 97 (2008) 4840
23. X. Qiu, J. W. Thompson, and S. J. L. Billinge, ’PDFgetX2: A GUI driven program to obtain the pair distribution function from X-ray powder diff raction data’, J. Appl. Cryst 37 (2004) 678 Copyright ⓒ International Union of Crystallography.
Author Biographies
Dr. Anne Kavanagh joined AstraZeneca in 2000, initially in the fi eld of solid state characterization, and more recently as a Crystallization Scientist. She is particularly interested in exploiting and tailoring the properties of the solid form, whether crystalline or amorphous, to achieve desirable properties, such as improved bioavailability, ease of handling, formulation and process robustness.
Dr. Helen Blade (né Hughes) is a Senior Scientist at AstraZeneca. She joined the company in 2007 having gained a Ph.D. with Professor Matt Rosseinsky at University of Liverpool, UK. Since then she has supported a range of early to mid-phase projects. Helen has been instrumental in the development of pairwise distribution function with AZ.
Dr. Jim McCabe Astrazeneca joined AstraZeneca Macclesfield 11 years ago, having previously worked for Merck Sharp and Dohme. His areas of interest include solid-state characterization of pharmaceutical materials, rational screening of solid forms (polymorphs, solvates and salts), crystallization strategies for poorly crystalline materials, structural aspects of crystallization from solution and stabilization of amorphous materials.
Dr. Ian McConvey is now an independent Process Consultant. For the past 31 years he has worked in industry and academia and is a Fellow of the Institution of Chemical Engineers. Whilst at AstraZeneca one of his activities was to support the early development of spray-drying technology and the subsequent externalization to contract manufacturers.
Dr. Steve Cosgrove has over 17 years experience in the solid state field and has applied this to a wide range of pharmaceutical projects from very early phase to late phase III. Over this time he has held a number of scientific and managerial positions relating to commercial development across the UK-US region of AstraZeneca.