David Jones, Sannova Analytical, VP Business Development
Meeting the Evolving Need for Effective Nitrosamine Detection
Resolving issues around nitrosamine (NA) contamination has demanded a complex and forensic investigation and the associated regulatory guidance continues to be refined, notably with respect to nitrosamine drug substance-related impurities (NDSRIs). In 2018, when unacceptable levels of N-nitrosodimethylamine (NDMA) were first found in the angiotensin receptor blocker (ARB) valsartan the initial thought was that this was an isolated case. Five years on, and at least 1400 recalled batches later, we know better.1
The intervening years have seen a swift and substantial response by regulators to safeguard patient health, and the rapid establishment of analytical methods and risk assessment frameworks to support pharmaceutical manufacturers. October 1st, 2023, was a key date, the deadline for the submission of changes to market authorizations associated with the detection and resolution of NA contamination issues,2 that marked the substantial progress made. Regulators and industrialists alike have stepped up, demonstrating some great science and skill. However, the resulting legacy is considerable: a much higher burden of trace analysis and an enhanced need for effective understanding, troubleshooting, and control, across the whole supply chain.
Here we consider what we now know about sources of NAs, the risk posed, and the challenges of detection. We present a validation criteria checklist for NA analysis and introduce an optimized single method for detecting a range of NAs of interest in a peptide drug. Validation data illustrate the excellent performance of this method with respect to accuracy, sensitivity, and linearity.
The Origin and Risk of Nitrosamine Contamination
Investigation of the NDMA contamination of valsartan revealed some salutary lessons. Figure 1 shows the change in the synthesis route made as valsartan transitioned to generic manufacturing. The switch from tributyltin azide to less expensive sodium azide necessitated the use of dimethylformamide (DMF) as a solvent, which was then recycled; NA formation was directly linked to DMF breakdown during the recovery and recycling process. The generic route was optimized for cost and long-term environmental impact - laudable aims - but the net effect was problematic and, at the time, not readily detectable due to the lack of appropriately sensitive analytical methods.
NAs are characterized by a nitroso group (N=O) bonded to an amine (NH2 ) and form via nitrosating reactions between amines and nitrous acid (which can be released by nitrite salts in acidic conditions – see Figure 2). ICH guidance for industry classifies NAs as a ‘cohort of concern’ because of their potential genotoxicity.3
Given that the processing conditions routinely used in active pharmaceutical ingredient (API) synthesis are highly suitable for NA formation, and the prevalence of amine sources across the supply chain, it soon became clear that the potential for NA contamination was far from isolated. In guidance issued in 2021 the FDA highlighted seven NAs that could theoretically be present in APIs or drug products, noting that five had already been detected.2
Sources of amines that may go on to form NAs include degradation products of the API itself; reagents, catalysts and solvents; contaminants in raw materials including water; recovered and recycled solvents; and packaging materials. The process of quenching is especially significant with respect to NA formation, and there is considerable potential for contaminants, once introduced into the supply chain, to persist and transition through to finished APIs and drug products. A key point to note is that NA contamination is not limited to chemically synthesized drug products; the developed regulations also apply to biologics.
From this starting point of seven compounds, the list of NAs of interest has progressively expanded. A crucial development has been the focus on NDSRIs as evidenced by more recent FDA guidance.4 NDSRIs, as the name suggests, arise from the nitrosation of amine groups on API molecules. They share structural similarities with a specific API, may be difficult to detect, and typically have unknown mutagenic/ carcinogenic properties. The new guidance helps manufacturers assess risk and set appropriate acceptable intake (AI) levels; high profile product withdrawals of Orphenadrine Citrate ER Tablets (Sandoz)5 and Quinapril HCl/Hydrochlorothiazide (Pfizer)6 evidence growing concerns in this area.
Assessing Regulatory Requirements for Detection and Control
The regulators have established AI levels for a substantial number of NAs, and for a range of NDSRIs, based on their potential for harm. These range from 26.5 ng/day for compounds of high potency to 1500 ng/day for those viewed as lower risk.4 It is worth noting that individual AI values are only applicable when a single NA is detected; with multiple NAs the total AI is 26.5 ng/day. The expectation is that manufacturers will implement effective controls to meet these levels, a far more demanding task for some NAs, and for some products than for others.
Let’s take a closer look at the relationship between AI levels, the maximum daily dose (MDD) and detection/analytical requirements. If we start with NDMA then this has an AI limit of 96 ng/day. We can determine what this means in terms of detection levels by dividing by the MDD. If patients take more of the drug, then we need to detect it at lower levels to avoid exceeding the AI, similarly if the AI is lower; N-nitrosodiethylamine (NDEA) another routinely detected NA has the lowest possible AI, just 26.5ng/day. For a MDD of 880mg or less the FDA indicates that a limit of quantification (LOQ) of 0.03 ppm is needed to meet the 26.5ng/day limit, but for an MDD of 1200 mg, for example, this figure falls to 0.02 ppm.2
The Challenges of Measurement
The LOQ requirements associated with NA detection are demanding but the task of meeting the new regulatory requirements is compounded by other factors. Since reliable analytical information for NAs, precursors and NDSRIs is the foundation of both product quality assurance and effective troubleshooting the quantity of trace analysis required is substantial and sufficient to test overall industry capacity. Substantial effort is required to confirm the safety of existing drugs, but NA detection must become routine early in the drug development process where there remains flexibility to modify reagents and processes to design out an issue. NA detection for new chemical entities is therefore a critical, ongoing task.
Against this backdrop, effectively leveraging in-house capacity is crucial but contract providers have a vital role to play, often holding the expertise required to develop, validate, and apply the required methods swiftly and efficiently. The regulators have released several analytical methods for NA detection but where methods need optimization for, for example, slightly different analytical systems, pharmaceutical manufacturers may lack the expertise and/or resources for assured application. In the current climate, the underutilization of any applicable analytical instrument is highly undesirable.
The first FDA method released - ‘GC/MS Headspace Method for Detection of NDMA in Valsartan Drug Substance and Drug Product’7 – highlights a key point about the methods being established which is that they are typically specific to both NA and drug. This method, for example, proved unsuitable for NDMA detection in ranitidine due to the degradation of ranitidine and an associated elevation of NDMA levels. The solution was to switch from GC (Gas Chromatography) to LC (Liquid Chromatography) as evidenced by the published LC/ HRMS (High-Resolution Mass Spectrometry) method.8 We now know that GC/MS, LC/MS, LC/MS/MS, and LC/HRMS (High-Resolution MS) all have a place in the NA detection toolkit. These techniques bring the orthogonality needed for accurate detection at very low levels in complex matrices addressing issues such as the presence of isobaric species - species with the same mass but different structures - a known complication with NDMA.
Establishing methods for the evolving range of analytes of interest, including NDSRIs, in all associated drugs, calls for the application of advanced methods that fully exploit the inherent attractions of these core techniques. NA contamination analyses are complicated by significant interferences in the chromatography step, excipients that are not easily extractable, and the very small masses of NAs involved. Our experience highlights the value of:
- Optimizing LC steps using appropriate reference standards to address solubility issues and tackle the problem of mass suppression from excipient species.
- Deploying advanced extraction techniques such as solid-phase microextraction or stir bar sorptive extraction to minimize sample loss, enhance recovery efficiency, and keep NAs intact for reliable detection.
- Robust atmospheric pressure chemical ionization (APCI) for mitigating the ionization effects associated with complex and variable sample matrices.
- Minimizing sample preparation, for example, via direct injection or direct sample analysis.
- Rigorously testing for robustness, for example, in the face of subtle changes to reagents and conditions, as may be required to optimize cost-efficiency and reduce environmental impact.
To date we’ve successfully tested over 170 drug substances and drug products, including for NDSRIs, notably relying on Triple Quadrupole MS (TQ/MS) and HRMS platforms for the flexibility and performance required to develop methods that are both fast and efficient; single methods for the detection of multiple NAs are a notable achievement. The validation criteria we find most useful are listed below and followed by a case study demonstrating method development and application.
Case Study: Screening for NAs in a Biological API with an Optimized TQ LC/MS/MS Method
Figure 3/Table 2 show linearity data for NDMA for a TQ LC/MS/MS method (AB Sciex API 6500) optimized to screen for a range of NAs in a peptide API. More specifically, the focus was to determine the presence or otherwise of ten specific NAs: NDMA, NDEA, NEIPA (N-Ethyl-N[1]nitroso-2-propanamine), NDIPA (N-Nitrosodiisopropylamine), NDBA (N-Nitrosodibutylamine), NMPA (N-Nitrosomethylphenylamine), NMBA (N-Nitroso-N-methyl-4-aminobutyric Acid), NMOR (N-Nitrosomorpholine), MNP (N-Methyl-4-Nitrosopiperazine) and NNV (N-nitroso-varenicline).
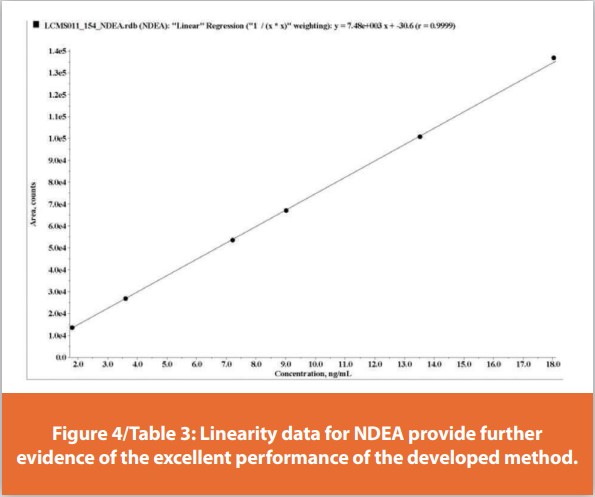
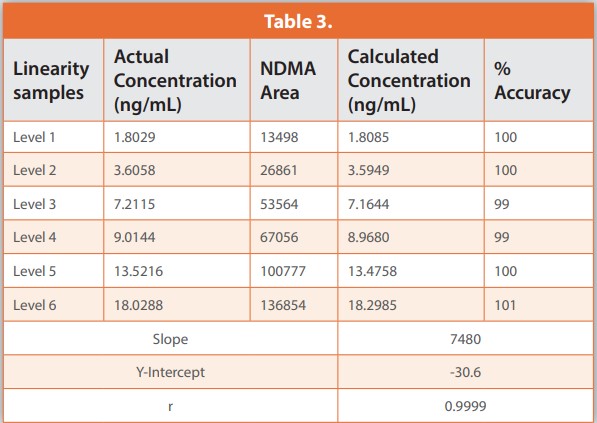
Linearity was assessed for each NA using a set of six combination calibration standards, each containing all ten NAs. The data show excellent linearity across the concentration range for NDMA which runs from 2 to 20 ng/ml. Accuracy is well within the acceptance criteria for each standard (80 – 120%), notably so for the most dilute, Level 1, Limit of Quantification (LOQ) solution; accuracy 102% c.f. a requirement of between 70 and 130%. The correlation coefficient for the linearity plot (r) is 0.9977 which also comfortably meets the acceptance criteria (r not less than 0.98). The gradient of the curve (slope 35000) indicates good sensitivity.
Figure 4/Table 3 shows equivalent data for NDEA demonstrating even better performance; accuracy ranging from 99 – 101% across all six calibration standards, an r-value of 0.9999. More broadly all linearity values were confirmed as meeting the established validation criteria for each of the ten NAs except a single accuracy point (74% accuracy for the Level 5 calibration solution for NMPA). In addition, the method was shown to robustly meet system precision and system suitability criteria (as defined above – data not shown). This represents exceptional analytical performance given the difficulty of establishing a single method for the detection of multiple, individual NAs.
Table 4 shows the screening data for three samples of the API along with the LOQ figures for each of the NAs which were established by converting the concentration of the measured Level 1 calibration standard into a ppm figure relative to the drug; Limit of Detection (LOD) values were determined from LOQ data using the signal-to-noise ratio method. The results confirm the absence of any of the ten NAs in any of the drug samples simultaneously demonstrating the utility and performance of the method for broad and robust NA screening.
Going Forward
That initial discovery of NAs in 2018 has led to demands for sufficiently sensitive detection that continue to evolve, testing both the skill and capacity of analytical laboratories. The case study presented here illustrates the abilities of contract providers who have an important role to play in meeting the growing need for trace analysis. The NA – drug specific nature of the analyses required, exacting LOQ requirements, and the sheer volume of testing required create a significant and ongoing challenge for the industry but there is considerable support, help, and capacity available for those that need it.
References
- S.S. Bharate ‘Critical Analysis of Drug Product Recalls due to Nitrosamine Impurities’ J. Med. Chem. 2021, 64, 6, 2923–2936.
- US Food and Drug Administration, Center for Drug Evaluation and Research, Guidance for Industry ‘Control of Nitrosamine Impurities in Human Drugs’ Rev 1, Feb 2021
- ICH guidance for industry M7(R1) Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk, July 2023.
- US Food and Drug Administration, Center for Drug Evaluation and Research, Guidance for Industry ‘Recommended Acceptable Intake Limits for Nitrosamine Drug Substance Related Impurities (NDSRIs)’ Aug 2023
- Company Announcement ‘Sandoz, Inc. Issues Nationwide Recall of 13 Lots of Orphenadrine Citrate 100mg Extended-Release Tablets Due to Presence of a Nitrosamine Impurity.’ March 22, 2022. Available to view at: https://www.fda.gov/safety/recalls-market-withdrawals-safety-alerts/sandoz-inc-issues-nationwide…
- Company Announcement ‘Pfizer Voluntary Nationwide Recall of Lots of ACCURETICTM (Quinapril HCl/Hydrochlorothiazide), Quinapril and Hydrochlorothiazide Tablets, and Quinapril HCl/Hydrochlorothiazide Tablets Due to N-Nitroso-Quinapril Content.’ March 22, 2022. Available to view at: https://www.fda.gov/safety/recalls-market-withdrawals-safety-alerts/Pfizer-voluntary-nationwide-recall-lots-accuretictm%E2%80%A6
- US Food and Drug Administration ‘GC/MS Headspace Method for Detection of NDMA in Valsartan Drug Substance and Drug Products’ Updated 25th Jan 2019
- US Food and Drug Administration ‘Liquid Chromatography-High Resolution Mass Spectrometry (LC-HRMS) Method for the Determination of NDMA in Ranitidine Drug Substance and Drug Product’ 13th Sep 2019
Publication Detail
This article appeared in American Pharmaceutical Review:Vol. 27, No. 1Jan/FebPages: 13-17
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special offers
from American Pharmaceutical Review – all delivered right to your inbox! Sign up now!