By: Sanjana Mareguddi, Dr Somashekar M Metri*, Agasa Ramu Mahesh
*Corresponding Author
Abstract
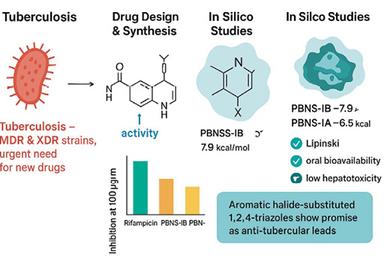
Tuberculosis (TB) is a significant worldwide medical problem aggravated by the advent of multidrug-resistant (MDR) and substantially drug-resistant (XDR) variants. This research concentrated on designing and synthesizing novel 1,2,4-triazole derivatives based on isoniazid, with the goal of identifying potential anti-tubercular agents. The newly synthesized derivatives, PBNS-IA and PBNS-IB, were assessed through their physicochemical properties, thin layer chromatography (TLC), melting point analysis, Fourier-transform infrared spectroscopy (FTIR), nuclear magnetic resonance (NMR), and mass spectrometry. In silico studies, including molecular docking against dihydrofolate reductase (PDB ID: 1DF7) and ADME/T predictions, were conducted to assess binding affinity and drug-like characteristics. PBNS-IB exhibited stronger binding interactions (–7.9 kcal/mol) than PBNS-IA (–6.5 kcal/mol), indicating favorable pharmacokinetic properties. The in vitro anti-tubercular activity was evaluated using the Microplate Alamar Blue Assay (MABA), which showed a dose-dependent inhibition of Mycobacterium tuberculosis. At a concentration of 100 μg/ml, PBNS-IB demonstrated greater inhibition (~56%) compared to PBNS-IA (~44%), although both were less effective than rifampicin (>80%). These results suggest that 1,2,4-triazole derivatives could be promising candidates for further development in anti-tubercular drug research.
KEYWORDS: 1,2,4-Triazole · Anti-tubercular activity · Molecular docking · ADME/T · Drug design · In vitro evaluation.
Introduction
Tuberculosis (TB) remains one of the most formidable contagious diseases worldwide, exhibiting higher death rates than HIV/AIDS. The World Health Organization (WHO) Global Tuberculosis Report (2020) indicates that nearly 10 million new TB cases were identified in 2019, with a substantial portion being multidrug-resistant (MDR) or resistant to rifampicin. The rise of MDR and extensively drug-resistant (XDR) TB strains has significantly hindered the efficacy of current treatment options, highlighting the pressing need for innovative chemical compounds that possess strong anti-tubercular properties.1,2
Among heterocyclic compounds, azoles constitute a significant category of nitrogen-containing five-membered aromatic heterocycles that are extensively utilized in the pharmaceutical industry. Within this category, the 1,2,4-triazole framework has garnered particular interest due to its capability to engage in various non-covalent interactions at the receptor binding site, functioning as both a hydrogen bond donor and acceptor. The polar characteristics of the triazole ring enhance its aqueous solubility, metabolic stability, and contribute to its advantageous pharmacological profiles. As a result, a variety of 1,2,4-triazole derivatives have been found to display an extensive series of biological deeds, including antifungal, antibacterial, antitumor, and particularly, antitubercular effects.3,4
In drug discovery, lipophilicity, solubility, and pKa (ionization) play significant roles in determining absorption, distribution, metabolism, excretion, and toxicity (ADMET), as illustrated in Figure 1. Quantitative structure - activity relationship (QSAR) studies further highlight the importance of balancing physicochemical properties to optimize activity and safety. For triazoles, structural modifications can significantly influence binding to molecular targets such as dihydrofolate reductase, thereby modulating anti-tubercular potential.5,6
Figure 1. Drug design
Several marketed drugs demonstrate the pharmacological importance of the triazole moiety, including ribavirin (antiviral), alprazolam (anxiolytic), rizatriptan (antimigraine), and letrozole (antitumoral). Isoniazid is a first-line anti-tubercular drug that undergoes activation by the catalase-peroxidase enzyme (KatG) in M. tuberculosis. The activated version inhibits InhA, an enoyl-acyl carrier protein reductase, which prevents the production of mycolic acids critical for the bacterial cell wall. This mechanism is illustrated in Figure 2. Furthermore, recent clinical evaluations of triazole-based anti-tubercular candidates, such as I-A09, highlight the potential of this scaffold in combating TB.6,7
Building on these insights, the present study focused on the design, synthesis, characterization, docking, ADME/T profiling, and in vitro assessment of novel 1,2,4-triazole derivatives (PBNS-IA and PBNS-IB) as possible anti-tubercular agents. The integration of synthetic chemistry, computational drug design, and biological evaluation provides a rational framework for identifying lead molecules with improved efficacy and drug-likeness.
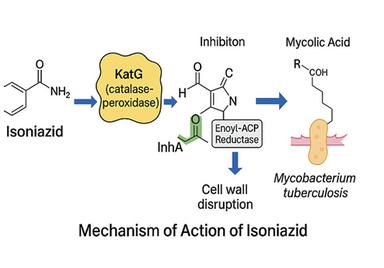
Figure 2. MOA of Isoniazid
Materials And Methods
Chemicals and Reagents
Isoniazid, carbon disulfide, potassium hydroxide, substituted aromatic halides, glacial acetic acid, and solvents (ethanol, methanol, hydrochloric acid, ammonia) were purchased from standard chemical suppliers. All reagents utilized were of analytical quality. Thin-layer chromatography (TLC) was accompanied on silica gel plates with suitable solvent systems, and melting points were measured using the open tube system.
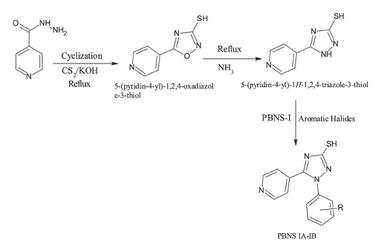
Scheme
Synthesis of 1,2,4-Triazole Derivatives
Synthesis of 5-(pyridine-4-yl)-4H-1,2,4-triazole-3-thiol
Isoniazid was refluxed with carbon disulfide in ethanolic potassium hydroxide solution for 3 hours. After the addition of a few drops of ammonia solution, the reaction mixture was acidified with concentrated hydrochloric acid. The precipitate obtained was filtered and dried. The product was monitored by TLC and characterized by its melting point.8
| Table 1. Physicochemical properties of 5-(pyridin-4-yl)-4H-1,2,4-triazole-3-thiol |
|---|
| SL.NO | Parameter | 5-(Pyridin-4-yl)-4H-1,2,4-triazole-3-thiol |
|---|
| 1 | Molecular Formula | C7H7N4S |
| 2 | Molecular Weight | 179 g/mol |
| 3 | Yield | 55.3% |
| 4 | Melting Point | 306 °C |
| 5 | Rf Value | 0.65 |
The intermediate compound was heated under reflux with a halogenated aromatic compound in the presence of glacial acetic acid for 5 hours. The reaction mixture was then poured over crushed ice, and the resulting precipitate was filtered and dried. TLC analysis verified the formation of the product.
| Table 2. Physicochemical properties of PBNS-IA |
|---|
| SL.NO | Parameter | PBNS-IA |
|---|
| 1 | Molecular Formula | C14H12N4S |
| 2 | Molecular Weight | 268.08 g/mol |
| 3 | Yield | 42.9% |
| 4 | Melting Point | 134 °C |
| 5 | Rf Value | 0.8 |
The intermediate was reacted with 2-chlorobenzyl chloride and glacial acetic acid under reflux for 5 hours. The reaction mixture was poured onto ice, and the product obtained was filtered, dried, and confirmed by TLC.
| Table 3. Physicochemical properties of PBNS-IB |
|---|
| SL.NO | Parameter | PBNS-IB |
|---|
| 1 | Molecular Formula | C14H12N4SCl |
| 2 | Molecular Weight | 302.04 g/mol |
| 3 | Yield | 43.7% |
| 4 | Melting Point | 134 °C |
| 5 | Rf Value | 0.5 |
Spectroscopic Characterization
PBNS-IA Light yellow: 42.9% 1H NMR (400 MHz, DMSO-d6): δ 8.80 (2H, d, J ≈ 8.6 Hz,), 7.80 (4H, m, 7.813, 7.809, 7.801, 7.797). Mass C14H12N4S m/z (%), calcd. (found): 268 (270.24, M-1).
PBNS-IB Yellow: 43.7% ¹H NMR (400 MHz, DMSO-d₆): δ 8.80 (2H, d, J = 8.7 Hz), 7.80 (2H, d, J = 8.0 Hz). Mass C14H12N4SCl m/z (%), calcd. (found): 302(304.25, M-1)
In Silico Studies
Molecular Docking
Molecular docking was executed via AutoDock Vina against dihydrofolate reductase (PDB ID: 1DF7). The overall docking process involved several key steps: ligand preparation, protein target selection, docking simulation, and analysis of the docked complex, as shown in Figure 3. The protein structure was retrieved from the RCSB PDB and prepared by eradicating water molecules and adding polar hydrogens. Ligand structures were generated in ChemSketch, converted into 3D conformations using Open Babel, and saved in PDBQT format. Docking simulations were carried out in AutoDock Vina, and the resulting docked complexes were evaluated based on binding energy and molecular interactions using PyMOL and Discovery Studio. The binding affinity of the Ligand and protein is shown in Table 4, and the Ligand-Protein interaction is shown in Figure 4, 5 & 6.
ADME/T Prediction
Drug-likeness and pharmacokinetic properties were predicted using Molinspiration and ADMETLab 3.0. Parameters such as molecular weight, hydrogen bond donors/acceptors, logP, solubility, gastrointestinal (GI) absorption, blood-brain barrier (BBB) penetration, and hepatotoxicity were evaluated.9,10
In Vitro Anti-tubercular Evaluation
Anti-tubercular activity was calculated using the Microplate Alamar Blue Assay (MABA). Mycobacterium tuberculosis H37Rv was cultured to mid-log phase. Test compounds (PBNS-IA and PBNS-IB) were evaluated at concentrations of 20–100 µg/ml. Rifampicin was used as the positive control, and DMSO as the negative control. After incubation at 37 °C for 6 days, 10% Alamar Blue solution was supplemented, and absorbance was measured at 600 nm. Percentage growth inhibition was calculated relative to the control.11,12
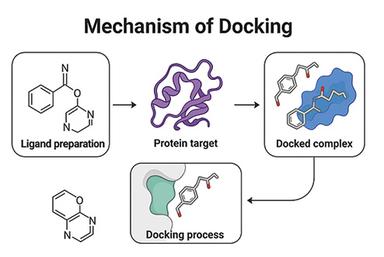
Figure 3. Mechanism of Docking
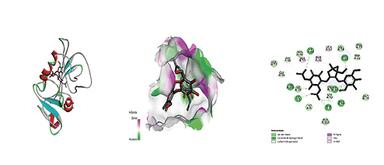
Figure 4. Interaction between ligand Streptomycin & Protein 1DF7
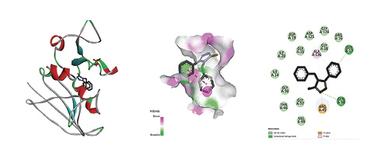
Figure 5. Interaction between ligand PBNS-IA & Protein 1DF7
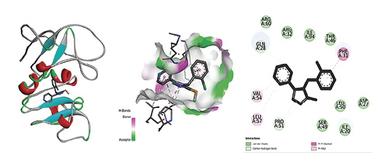
Figure 6. Interaction between ligand PBNS-IB & Protein 1DF7
Results And Discussion
Synthesis of 1,2,4-Triazole Derivatives
Two novel 1,2,4-triazole derivatives (PBNS-IA and PBNS-IB) were synthesized from isoniazid through a three-step reaction pathway. The yields were 42–55%, and TLC and melting point analysis established the purity of intermediates and final products. The successful synthesis demonstrated the feasibility of designing triazole derivatives as new anti-tubercular scaffolds.
Spectroscopic Characterization
FTIR, NMR, and mass spectrometry confirmed the structures of the synthesized compounds.
- PBNS-IA exhibited –NH stretching at 3574 cm-¹, aromatic C–H peaks at 2981 cm-¹, and an M+ peak at m/z 270.24, reliable with its expected molecular weight.
- PBNS-IB showed a characteristic –Cl peak at 826 cm-¹ and a molecular ion peak at m/z 304.25.
The spectroscopic data supported the successful introduction of substituents and confirmed structural integrity.
Molecular Docking Studies
Molecular docking against dihydrofolate reductase (PDB ID: 1DF7) revealed that PBNS-IB (–7.9 kcal/mol) displayed better binding affinity than PBNS-IA (–6.5 kcal/mol), though slightly weaker than the standard drug streptomycin (–8.8 kcal/mol). PBNS-IB formed interactions with key residues (Ile20, Leu57, Arg32, Phe31), suggesting its potential as a competitive inhibitor. The enhanced binding of PBNS-IB may be attributed to the chloro-substituted benzyl group, which increases hydrophobic interactions at the binding site.
ADME/T Prediction
ADMET analysis indicated that both PBNS-IA and PBNS-IB complied with Lipinski’s rule of five, demonstrating good drug-likeness. The predicted solubility, moderate gastrointestinal absorption, and low hepatotoxicity risk suggest acceptable pharmacokinetic profiles. PBNS-IB exhibited slightly better oral bioavailability predictions compared to PBNS-IA, which aligns with its higher binding affinity in docking studies.
Table 5. ADMET study |
|---|
SI NO | Compound code | MW | HD | HA | Wlog P | ESol | BBB | GI | Bioavailability | TPSA | Hepatotoxicity |
|---|
01 | Streptomycin | 581.27 | 16 | 19 | -1.903 | -0.936 | | | 0.17 | 336.43 | 0.904 |
02 | PBNS – IA | 268.08 | 0.0 | 4.0 | 1.855 | -3.162 | 0.026 | 0.149 | 0.406 | 43.6 | 0.744 |
03 | PBNS-IB | 302.04 | 0.0 | 4.0 | 2.35 | -3.692 | 0.027 | 0.148 | 0.542 | 43.6 | 0.764 |
In Vitro Anti-tubercular Evaluation
Both compounds were evaluated for their inhibitory effects against Mycobacterium tuberculosis using the Microplate Alamar Blue Assay. The inhibition was dose-dependent:
- PBNS-IA achieved a maximum inhibition of 43.9% at 100 µg/ml.
- PBNS-IB showed stronger activity, with 56.7% inhibition at 100 µg/ml.
- Rifampicin, used as the standard, demonstrated >80% inhibition at the same concentration.
These findings confirm that structural variations significantly influence biological activity. PBNS-IB consistently outperformed PBNS-IA across docking, ADME/T predictions, and in vitro assays, highlighting the potential of halogen-substituted triazole derivatives as promising leads for anti-tubercular drug development.
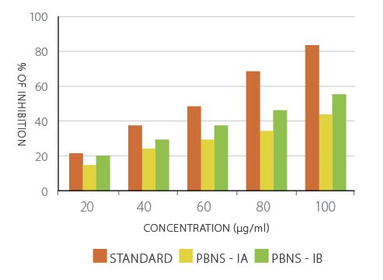
Graph No.1. Anti-TB Activity of Standard and PBNs
Conclusion
Figure 7. Anti-TB Assay of Standard and PBNs in 96-Well Plate
In this study, two novel 1,2,4-triazole derivatives (PBNS-IA and PBNS-IB) were successfully synthesized from isoniazid and characterized using TLC, melting point, FTIR, NMR, and mass spectrometry. Molecular docking studies against dihydrofolate reductase (PDB ID: 1DF7) revealed favorable binding affinities, with PBNS-IB (–7.9 kcal/mol) showing stronger interactions than PBNS-IA (–6.5kcal/mol).
ADME/T predictions indicated that both compounds possessed acceptable drug-like properties with moderate oral bioavailability and low hepatotoxicity risk. In vitro anti-tubercular activity, evaluated using the Microplate Alamar Blue Assay, demonstrated dose-dependent inhibition of Mycobacterium tuberculosis, with PBNS-IB exhibiting greater activity (~57% inhibition at 100 µg/ml) compared to PBNS-IA (~44%). Although both were less potent than rifampicin, the findings highlight the potential of halogen-substituted triazole derivatives as promising scaffolds for anti-tubercular drug discovery. Further structural optimization and biological evaluation may enhance their efficacy and overlay the approach for the improvement of innovative therapeutic substitutions.
Funding
This work was done without any external support.
Competing Interest Policy
The authors declare no competing interests.
Ethics, Consent To Participate, And Consent To Publish Declarations
Not applicable.
Table 6: Anti-TB Assay of derivatives PBNS-IA & PBNS-IB |
|---|
SI NO | SAMPLE CODE | Concentration (µg/ ml) | Absorbance at 600nm | Mean | % Of Growth Inhibition |
|---|
Test 1 | Test 2 | Test 3 |
|---|
1 | Growth C | | 1.731 | 1.731 | 1.731 | 1.731 | |
2 | Standard Rifampicin | 20 | 1.353 | 1.352 | 1.353 | 1.353 | 21.88% |
| | | 40 | 1.078 | 1.075 | 1.079 | 1.077 | 37.81% |
| | | 60 | 0.888 | 0.886 | 0.89 | 0.888 | 48.72% |
| | | 80 | 0.534 | 0.532 | 0.536 | 0.534 | 69.16% |
| | | 100 | 0.265 | 0.267 | 0.263 | 0.265 | 84.69% |
3 | PBNS-IA | 20 | 1.455 | 1.45 | 1.458 | 1.454 | 16.00% |
| | | 40 | 0.309 | 1.305 | 1.312 | 1.308 | 24.43% |
| | | 60 | 1.21 | 1.208 | 1.216 | 1.211 | 30.04% |
| | | 80 | 1.1 | 1.098 | 1.113 | 1.103 | 34.71% |
| | | 100 | 0.97 | 0.968 | 0.972 | 0.97 | 43.96% |
4 | PBNS-IB | 20 | 1.38 | 1.376 | 1.383 | 1.379 | 20.33% |
| | | 40 | 1.225 | 1.221 | 1.227 | 1.224 | 29.28% |
| | | 60 | 1.065 | 1.063 | 1.068 | 1.605 | 38.47% |
| | | 80 | 0.925 | 0.92 | 0.93 | 0.925 | 46.56% |
| | | 100 | 0.75 | 0.748 | 0.753 | 0.75 | 56.67% |
Acknowledgment
We acknowledge the management of BLDEA’s Shri Sanganabasava Mahaswamiji College of Pharmacy and Research Centre, Vijayapura, for providing the infrastructure and support for this study.
About the Authors
Ms. Sanjana Mareguddi (B. Pharm) is a postgraduate student in the Department of Pharmaceutical Chemistry at BLDEA’s Shri Sanganabasava Mahaswamiji College of Pharmacy and Research Centre, Vijayapura, Karnataka. She possesses expertise in in silico studies, including molecular docking, ADMET prediction, and visualisation. She is proficient in handling various laboratory instruments and conducting synthetic reactions, thin-layer chromatography (TLC), and plant extraction techniques. She has participated in three workshops on computer-aided drug design and two scientific conferences. She has reviewed one article in a national journal under the UGC Care list and has one published patent and one book chapter to her credit. Additionally, she has submitted four review articles and two research articles for publication in reputed journals.
Dr. Somashekhar M Metri (M.pharm , Ph.D.) , is currently serving as an Associate Professor and HoD in department of pharmaceutical chemistry at BLDEA’s Shree Sanganabasava Mahaswamiji College of Pharmacy and Research Centre, Vijayapura, Karnataka. With over 13 years of teaching and research experience. He has specialized in the area of drug design and docking, synthetic chemistry, 2D-QSAR study, study of SAR of synthesized compounds, handling of analytical instruments, computer aided drug design study and analysis of pharmacological activities of newly synthesized compounds. He works as a reviewer for more than 20 various peer reviewed national and international journal like WoS, Scopus, and Elsevier journals. Organized more than 12 conferences and seminars as secretory and participated in more than 55 various national and international seminars, conferences and faculty development programs . Appointed as Benthamscience Journal Ambassadors year 2019-20.
Dr. Agasa Ramu Mahesh is a Professor and Head of the Department of Pharmaceutical Chemistry at The Oxford College of Pharmacy, Bengaluru. He holds a PhD in Pharmaceutical Chemistry and has extensive academic and research experience in drug design and development. His research interests include antidiabetic, anticancer, and anti-Alzheimer’s drug discovery, as well as drug repurposing strategies. He has published scholarly work in these areas and actively mentors undergraduate and postgraduate research. His expertise spans medicinal chemistry, computational drug discovery, and pharmaceutical research methodologies.
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special
offers from American Pharmaceutical Review delivered to your inbox!
Sign up now!