Introduction
For many sterile products, preparation of the patient’s dose in a hospital setting requires breaching the container and closure system long before the dose is administered. Examples of such products include solids which are constituted with a diluent, and admixtures of liquid products.
Recently, the Center for Drug Evaluation and Research (CDER) has reviewed several New Drug Applications (NDAs) for nonpreserved products intended for final preparation and storage in a hospital pharmacy prior to patient administration. The types of these products vary widely with regard to drug product family and diluent(s), as well as proposed holding times and conditions. When preparing these types of drug products, the hospital pharmacist relies on the product label to provide the following information related to the drug product’s final preparation and storage:
• The process of penetration(s) of the container (e.g.: needle gauge).
• The choice of diluent(s).
• The diluent volume.
• The temperature(s) at which the final product may be stored prior to administration.
• The duration(s) for which the final product may be stored prior to administration.
Following is an example of selected sections from the Dosage and Administration portion of a redacted product label. This example is included here to illustrate the use in this paper of the phrase “final drug product preparation and holding/storage conditions”.
⇒Preparation of Solutions
Product A does not contain a bacteriostatic preservative. Aseptic technique must be followed in preparation of the infusion solution.
Preparation of the 500 mg dose:
- Constitute the vial with 10 mL of sterile water for injection or 0.9% sodium chloride injection (normal saline) and gently shake to form a suspension.
- Withdraw the suspension using a syringe with a 21 gauge needle and add it to an infusion bag containing 100 mL of normal saline or 5% dextrose; gently shake until clear. The final infusion solution concentration is 4.5 mg/mL.
⇒Storage of Constituted Solutions
Upon constitution with sterile water for injection or 0.9% sodium chloride (normal saline) injection, the suspension in the vial may be held for 1-hour prior to transfer and dilution in the infusion bag.
Following dilution of the suspension with normal saline or 5% dextrose, infusions stored at controlled room temperature or under refrigeration should be completed according to the time in the following table.

At CDER, there is an expectation that the above information in the proposed product label be accompanied by supporting information in the NDA. If this information is not in the NDA, then we request that it be supplied in an amendment to the application for review. Recently, there has been an expression of dissatisfaction on the part of some applicants regarding our request for this information. Applicants have wanted explanations as to why we need this information and whether there is guidance on the subject. The purpose of this paper is to address these questions and to summarize the information that an applicant should provide in a submission to FDA in support of the product’s post preparation holding time and conditions. Additionally, microbiological data collected during pharmaceutical product development will ultimately translate into the product label information summarized above.
Historical Overview
Pharmaceutical companies go to great lengths to manufacture a drug product which is sterile, whether by terminal sterilization or aseptic processing. Equally important to the microbiological quality of the product at the time of product release is its microbiological quality at the time of patient administration. Many sterile products are administered immediately after penetration of the container closure system. For these types of products, there is no time for growth of any microorganisms that may contaminate the product during preparation of the dosage prior to administration. In contrast to these immediate-use injectable products, there are sterile, non-preserved drug products which are penetrated in a hospital pharmacy for the purpose of final product preparation, held for a period of time, and then administered to the patient. For these types of products, patient safety is at risk if adventitious microorganisms are capable of growing in the final drug product during the holding period.
Once a drug product’s container has been breached, it is assumed that microbial contamination may have occurred. This point is illustrated by the fact that the use of multiple dose unit vials has contributed to nosocomial infection outbreaks (4). Studies of multiple dose unit use provide evidence that penetration of a sterile product followed by its storage and later administration to the patient increases patient risks of infection as compared to single use products which are administered immediately following product penetration (6). If we therefore assume that container penetration may result in microbial contamination of the drug product, then the microbiological quality of the final product at the time of patient administration is determined by the final product’s ability to support microbial growth under the conditions of the storage period. Clearly, all drug products differ with respect to their ability to support microbial growth. As an example, investigators of nosocomial infection outbreaks associated with the administration of the lipid-based propofol concluded the following regarding the correlation between this drug’s ability to support microbial growth and the resultant patient infections (5):
“To prevent further outbreaks, the people administering the agents must fully understand the ability of these drugs to support microbial growth so as not to put the patients at risk.”
To summarize thus far, we assume that container penetration may result in microbial contamination of the drug product, and we understand that some drug products are conducive to microbial growth. As a result of these two points, those of us on CDER’s New Drug Microbiology Staff believe that finished product storage conditions and related holding periods should be product specific and supported by scientific data. Additionally, ICH guidance documents are quite clear regarding this subject. ICH Q8 Pharmaceutical Development states the following in Microbial Attributes, Section 2.5 (2):
“Where relevant, microbial challenge testing under testing conditions that, as far as possible, simulate patient use should be performed during development and documented in this section.”
Further, ICH Q1A (R2) Stability Testing of New Drug Substances and Products provides the following information (1):
“Stability testing of the drug product after constitution or dilution, if applicable, should be conducted to provide information for the labeling on the preparation, storage condition, and in-use period of the constituted or diluted product. This testing should be performed on the constituted or diluted product through the proposed in-use period on primary batches as part of the formal stability studies at initial and final time points, and ...”
Finally, the following statement from ICH Q9 Quality Risk Management is extremely relevant to this subject (3):
“It is important to understand that product quality should be maintained throughout the product lifecycle such that the attributes that are important to the quality of the drug product remain consistent with those used in the clinical studies.”
Risk Assessment
When submitting applications for drug products in which the proposed product label identifies a storage period and holding conditions following initial container penetration, we suggest that the application include a risk assessment report which addresses the proposed holding period. To be clear, the risk assessment for these non-preserved products should not be confused with demonstration of the efficacy of a preservative system in a multiple dose use product. Preservative efficacy of a drug product is demonstrated by testing the drug product according to USP<51> (7). In contrast, the risk assessment to be used in support of the holding periods for those types of products described in this paper should consist of the following:
⇒A short summary evaluating the constituted product’s formulation with regard to its potential to support microbial growth. For example, does the final product possess antimicrobial activity? Will the product be constituted using a diluent containing a sugar or other microbial growth promoting components?
⇒Studies demonstrating that the product does not support adventitious microbial growth under the storage conditions. These studies are meant to demonstrate whether the product will allow an increase in microbial numbers, as compared to the USP<51> studies which demonstrate whether a product reduces a microbial challenge.
Microbial Challenge Study Design
Applicants should follow the guidelines below when designing the product growth promotion studies. We suggest that the challenge microbes include the panel provided in USP<51>, as well as typical skin microflora and nosocomial agents to simulate the types of flora that may contaminate a drug product in a hospital pharmacy. The final product should be inoculated with small numbers of each challenge microbe in individual containers. A small inoculum will simulate the level of possible contamination during penetration. Although the inoculum size should be small, it should also be both measurable and repeatable. For example, if a membrane filtration method is used for enumeration of the challenge organism, an inoculum size of less than 100 CFU/mL of product is appropriate. Following inoculation of the final product with the challenge organisms, the test units should be stored at the temperature(s) described in the proposed label. Samples should be removed periodically throughout the duration of the study for determination of microbial count for a period of time exceeding 2-3 times that of the maximum holding time in the proposed label. For maximum holding periods which are equal to or exceed one week, microbial challenge data extending 2 times that of the maximum hold time may be appropriate. In these cases, applicants are encouraged to contact CDER Review Microbiologists prior to the initiation of the challenge studies. The data derived from these studies may be presented in tabular formats, as well as in graphs. Applicants should be sure to include the raw data obtained from these studies, in addition to data summaries.
Determination of Microbial Growth
An important aspect of the microbial challenge studies is how best to interpret whether the challenge organisms have grown during the study. We have had much discussion regarding this topic within our review group here in CDER. The following statement regarding growth is taken from USP<51> (7): “No increase is defined as not more than 0.5 log10 unit higher than the previous value measured.” Although the USP<51> definition is useful when estimating baseline population counts, we believe that the observation of data trending is important in its interpretation. Following is an example of this point. The following table contains data obtained from a hypothetical microbial challenge experiment where the inoculum is less than 100 CFU/mL, and the requested maximum hold time is equivalent to Time Point #4.

Note that I have specifically chosen data for which there is “no increase” according to the USP definition with regard to comparison of Time Point 4 with Time Point 3, comparison of Time Point 5 with Time Point 4, and comparison of Time Point 4 with Time Point 1. A semi logarithmic graph of CFU/mL vs. Time of these data is illustrated below, showing the applicant’s requested maximum hold time equivalent to Time Point 4.
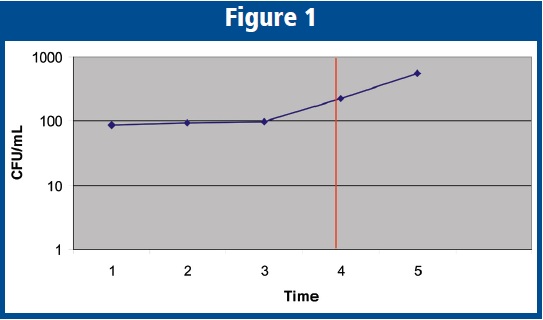
The reader should note that Time Point 4 conforms to the USP<51> definition of “no increase” when compared to the original Time Point 1. However, when comparing the difference between Time Point 5 with the original inoculum, clearly the challenge organism has “increased” or demonstrated growth, which also suggests that the organism has begun to show growth at Time Point 4, even though Time Point 4 conforms to the USP<51> definition. As a result, a maximum hold time equivalent to that of Time Point 4 would pose potential risk to the microbiological quality of the final product, and therefore would not be supported by this reviewer. The reader should also note that if the experiment were concluded at Time Point 4 (the maximum requested hold time), the ability to predict the trend of the data would have been lost. As presented in the graphic, the growth trend appears to signal the start of log-phase growth, which could occur earlier or later with different strains of a given species. Such growth would produce exponential increases in the microbial population that pose significant risk to patients. This concern is the reason for the suggested sampling frequency when determining microbial concentration: “Samples should be removed periodically throughout the duration of the study for determination of microbial count for a period of time exceeding 2-3 times that of the maximum holding time in the proposed label.” The reason that we suggest extending the experiment beyond the maximum requested time point is so that the data may be examined for trending and to establish a comfort level regarding the approved maximum storage time as stated in the product label.
Conclusion
The use of multiple dose unit drug dosage forms has proven that penetration of the container closure system may result in microbial contamination of the drug product. Further, our knowledge of the ability of microorganisms to increase in number at a rapid rate reminds us that if the drug product is conducive to microbial growth the microbiological quality of the contaminated product may be diminished in a very short period of time. Therefore, we believe that finished product storage conditions and related holding periods should be product specific and supported by scientific data. The risk assessment described in this paper is the mechanism that a pharmaceutical company should use to demonstrate that the preparation and storage conditions which are described in the product label do not put the final drug product at significant risk to be microbiologically unsafe to the patient.
References
1. Guidance for Industry, Q1A (R2) Stability Testing of New Drug Substances and Products. ICH. November 2003.
2. Guidance for Industry, Q8 Pharmaceutical Development. ICH. May 2006.
3. Guidance for Industry, Q9 Quality Risk Management. ICH. June 2006.
4. Mattner, F. & P. Gastmeier. 2004. Bacterial Contamination of Multiple-Dose Vials: A Prevalence Study. Assoc. for Professionals in Infection Control and Epidemiology. 32: 12-16.
5. Nichols, RL. & J.W. Smith. 1995. Bacterial Contamination of an Anesthetic Agent. New England Journal of Medicine. 333: 184-185.
6. Nogler-Semenitz. et. al. 2007. Bacterial Contamination of Solutions for Parenteral Administration for Single- and Multiple-dose Vials after Multiple Use in the Hospital. Wien med Wochesnschr. 157: 398-401.
7. USP 31/NF 26. United States Pharmacopeial Convention. 2008.
Dr. Metcalfe holds a BA in biology from Merrimack College in Andover, MA, and a PhD. in microbiology from the University of Rhode Island. Following his graduate studies, he worked as a college professor for a total of 14 years at Potsdam College of the State University of New York and Salem State College in Massachusetts where he taught microbiology related courses. During summers, John worked in several government (Natick Army Research Laboratories) and industry (Virus Research Institute and Avant Immunotherapeutics, Inc.) laboratories as a research microbiologist. John began his career at CDER in January of 2003, where he works as a review microbiologist.
To correspond with the author, please email him at: [email protected]
To read more on Microbiology, please visit our website (americanpharmaceuticalreview.com) and type “Microbiology” in the advanced search box