Abstract
Development of a successful subcutaneous antibody therapy requiring chronic administration at high dosing (several mg/kg), particularly when coupled with prefilled syringe and autoinjector device for at-home administration, is dependent on the development of a stable formulation at high concentration. This is very challenging due to a few key factors. First is the current limitation on subcutaneous injection volume (≤1.5 ml), and more importantly due to limited intrinsic stability in a highly concentrated aqueous solution. To overcome the instability barrier of the aqueous antibody solution, products are typically stored in a lyophilized (freeze-dried) state. Besides stability, the development of room temperature products, improved patient compliance and easier worldwide distribution are additional advantages of lyophilization. Unfortunately, the lyophilization process also generates a variety of freezing and drying stresses. Additionally, other factors such as a sub-optimal drying process, limited long-term storage stability and slow reconstitution time could influence the choice between a liquid and a lyophilized formulation. The purpose of this review is to discuss the scientific foundation and the critical issues related to the lyophilization process for development of stable antibody-based formulations. Additionally, some guidelines for successful development of lyophilized high concentration antibody formulations are presented.
Introduction
Since the generation of monoclonal antibody (mAb) through somatic cell hybrids in 1975 [1], antibody therapy has taken scientists, physicians and patients from early promises through the disappointment of failure, to beneficial products to the end user. The clinical possibilities of antibody therapy are obvious, with over 26 approved mAbs or related products for use in various indications including oncology, infectious diseases, rheumatic disorders and organ transplant and over 150 antibodies in clinical trials [2,3]. One of the main reasons that the antibody market is one of the largest and fastest growing amongst the biopharmaceuticals is the high approval rate (~ 21%) for mAbs by FDA (Food and Drug Administration) compared to only 5% for new chemical entities (NCEs) [4]. This higher success of antibody therapy over the conventional small molecule drugs stems from (a) high specificity of mAbs for their target and (b) well-characterized functional domain that can be manipulated to control antibody properties, (c) long half-life in the range of approx. 10-21 days that allows for a dosing regimen of approx. 7-30 days, and (d) technological advances in antibody selection and production. Looking toward the future, particularly with the race for biosimilars, the next-generation antibody formulations are focused on enhancing the clinical utility or application of the therapy with increased patient compliance.
Subcutaneous delivery over the conventional intravenous delivery, for example, is more appealing for frequent and chronic administration particularly when coupled with prefilled syringe and autoinjector device for at-home administration and improved compliance. Success of such formulations, however, hinges on their use at high concentration (> 100mg/mL) to allow efficacious does of several mpk (mg/kg) to be achieved due to the allowable administration volume limit of (<1.5 ml). However, due to limited pharmaceutical stability at high concentrations, significant formulation challenges exist. In general, while a liquid antibody formulation is generally more desirable from a marketing and manufacturing perspective because they are faster to develop and easier to prepare for administration than alternative formulation approaches, liquid antibody formulations are more prone to chemical and physical degradation pathways, such as oxidation, deamidation, aspartate isomerization, peptide bond hydrolysis and aggregation. The degradation pathways are hydrolytically driven. Water, for example, mediates the electron transfer during deamidation and oxidation event. Similarly, required addition of water across a peptide bond can be a critical step in peptide bond hydrolysis. In contrast to chemical degradation which generally is not concentration dependent, aggregation is generally the primary degradation pathway in high concentration antibody formulation, as aggregation requires bi-molecular collision and usually is concentration dependent. It should be noted, however, that the size of the aggregate, nature (reversible vs. irreversible) and the mechanism of aggregation (covalent vs. non-covalent) determines the relationship of concentration to antibody formation [5,6].
In one approach, aggregation at a given storage condition can be mitigated by using preferentially excluded osmolytes such as sucrose. At high antibody concentration, however, the mass of the stabilizing excipient needed to reach ratios similar to those at dilute antibody concentration can result in a highly viscous and/or hyperosmotic solutions with practical constraints on manufacturing, analytical, delivery and formulation. Another approach to overcome the instability barrier of the aqueous antibody solution is to store the product in a dry state as the long-term stability of biopharmaceuticals is generally enhanced when stored in a dried state than a liquid form. Lyophilization (freeze-drying), in this regard, is the most widely used approach for preparing solid antibody pharmaceuticals to achieve an acceptable shelf-life. Besides minimizing hydrolytic reactions, restricted mobility and/or conformational flexibility in presence of appropriate excipients are some of the additional benefits of lyophilization.
Considerations for Liquid vs. Lyophilized Formulation
The advantages of stability, storage, and ease of shipping make lyophilization the method of choice over a liquid formulation in early development, and potentially for the marketed product. The lyophilization process is a trusted aseptic process operation meeting the product sterility assurance requirement without the stress of terminal sterilization. Lyophilization also allows for attaining high concentration of antibody post reconstitution (by using a smaller volume of diluent) which is usually difficult to achieve through conventional methods. Additional factors such as the potential for a room temperature product, easier feasibility for at-home administration, the ability to deliver high doses (> 1 mg/ kg) in a minimal volume, resulting in increased patient compliance makes lyophilization a powerful approach to obtain commercially viable high concentration antibody formulation. Unfortunately, the freeze-drying process also generates a variety of freezing and drying stress. Additionally, lyophilization is a time- and energy- consuming process that requires a complex and lengthy unit operation resulting in higher cost and large capital investment. However at high concentration (>100 mg/ml), unrecoverable product loss ( as high as 20% due to adherence to container surface) can be easily minimized in a lyophilized formulation by using smaller vial or prefilled syringes where the plunger head pushes the solution off the surface thereby mitigating the increased cost without compromising the stability. Additional cost saving advantages of lyophilized formulations include aseptic bulk drying process with decreasing bulk storage space and removal of the need for cold chain with a room temperature product. Other factors such as sub-optimal drying process, limited long-term storage and slow reconstitution times could influence the choice between a liquid or lyophilized formulation. A significant advantage to the lyophilized state, specifically early in development, would be obvious by utilizing lyophilization for an early entry into clinical trials (e.g. safety assessment, phase I etc.) thereby allowing more time and resources dedicated towards optimizing the desired formulation (liquid vs. lyophilized) for a commercially desirable product. This review discusses the scientific foundation and the critical issues related to the lyophilization process for development of stable antibody-based formulations. Specifically, the interrelated process of lyophilization, the associated denaturation stresses and more importantly, its impact in formulating solid antibody formulations are discussed. Additionally, guidelines for successful development of a lyophilized high concentration antibody formulation will be presented.
Lyophilized Formulation Development
An optimal lyophilization process can be designed once the critical properties of the formulation, including but not limited to, glass transition temperature (i.e. softening temperature of amorphous systems designated as Tg’, Tg), collapse temperature (Tc), the properties of excipients used, stability and/or solubility of drugs are known to the development scientist. Given each antibody has a unique personality related to its stability requirement and the physical stability of monoclonal can be lowered, though not always, at high concentration due to crowding effects [7,8,9]; the stability issue is particularly important for formulating high concentration antibody formulations [10]. It should be noted that formulating antibodies, even at low concentration, present generic issues associated with protein therapeutics due to their proteinaceous nature although several other unique issues such as hinge region cleavage, glycation of lys residue, partial heavy chain C-terminal Lys processing, Fc glycosylation etc. are also present [11,12]. In addition to these issues, the freeze-drying process generates a variety of stresses such as potential pH changes, formation of ice crystals, solute concentration, cold-denaturation of antibody etc [13,14,15]. These issues are often mitigated by proper choice of formulation component and process parameters and therefore lyophilization development is approached in two inter-dependable phases namely, (a) optimization of formulation and (b) optimization of lyophilization cycle, respectively [10,16,17,18,19,20].
A typical freeze-drying cycle is comprised of three distinct stages: freezing, primary drying and secondary drying [21]. Development of a successful lyophilized product requires understanding of all three stages, the stresses associated with each and how to resolve the commonly encountered issues. These issues are discussed below with specific examples from the literature. This article will focus on understanding the significance of these aspects to avoid the commonly encountered freezing and drying stresses during lyophilization and storage condition.
Freezing
Freezing, the first step in lyophilization, is carried out at temperatures below Tg’ (glass transition temperature) for an amorphous or below Teu (eutectic temperature) for a crystalline state for sufficient period of time to allow for transformation of all solution into solid [21,22,23]. At the end of freezing, the water present is converted into ice and typically less than 20% of water (w/w) is in the non-ice state containing the concentrated solute glass. Additionally, the rate of cooling determines the size of ice crystals and the cake structure. Slow freezing, for example, usually results in formation of porous cake with larger ice crystals. However, slow freezing would also increase the freezing time and the time antibody stays in the concentrated phase. Additionally, an annealing step may be incorporated into the freezing step to allow efficient crystallization of the crystalline bulking agent (such as glycine and mannitol). To achieve high crystallization rate and complete crystallization, the annealing temperature is usually held between the Tg’ of the amorphous phase and Teu of the bulking agent. Crystallization of bulking agent such as mannitol, however, during the storage condition might compromise the stability of the product while during primary drying might result in vial breakage [24,25]. The optimal time required for the annealing step is dependent on the type of bulking agent used and the mass ratio of bulking agent to other solutes. It should be noted that annealing above Tg’ results in growth of ice crystals thereby decreasing the product resistance to the flow of water vapor eventually resulting in shorter primary drying times [26]. The product specific surface area, however, is reduced which may result in increased residual moisture content in secondary drying stage or may require a longer secondary drying [27].
The significance of freezing cannot be overemphasized due to the destabilizing stresses induced in this step, particularly for the proteinaceous drugs. These stresses include low temperature resulting in cold denaturation, increase in protein concentration leading to aggregation, surface-induced unfolding at the icewater interface, pH changes, phase separation, dehydration stress and increase in ionic strength [15,28]. Freezing-induced denaturation of proteins and antibody is a complex function of both temperature as well as cooling rate [29]. For example, it is known that antibodies are typically robust to freeze/thaw cycles, however, some cryoimmunoglobulins demonstrate the propensity to undergo precipitation as they are cooled from 37oC [30]. In contrast to the entropically driven thermal denaturation process, cold denaturation is an enthalpy driven process in which the solvophobic interactions decreases with decreasing temperature ultimately decreasing the stability [31,32]. Similarly, cooling rates and the degree of supercooling can also induce formation of aggregates by impacting the ice morphology [21]. For example, the propensity for aggregation was much higher at a faster cooling rate for bovine and human IgG compared to a slow freezing rate; an observation that could be attributed to the formation of smaller ice crystals with larger ice-water interface at faster freezing rate ultimately resulting in greater surface-induced unfolding and aggregation [18]. The degradation events at ice-aqueous interfaces can also be mitigated either by using surfactant or by saturating the protein-ice interface by increasing the antibody concentration [19]. A slow freezing rate (< 0.5oC/min), although favorable in the case discussed here, might damage a high concentration antibody formulation prone to phase separation as phase separation is a kinetically slow process. Additionally, at slow freezing, higher degradation might be observed as the rate of chemical reactions may be accelerated in partially frozen state due to many-fold increase in antibody concentration [33]. Freezing rate and duration may also influence type and extent of crystallization of bulking agent, such as mannitol, or buffer species that might result in pH shifts during freezing leading to poor stability or reconstitution times [33,34,35,36]. For example, the pH of sodium phosphate shifts from 7.5 to 4.1 upon freezing due to crystallization of basic buffer component (Na2HPO4.2H2O) while a modest increase in pH of 0.8 unit is observed for potassium phosphate [15,21]. Nucleation and subsequent crystallization of the buffer species may be prevented by using fast cooling rate [33]. Given the issues discussed above with fast and slow cooling rates, usually a moderate cooling rate of 1oC/min is preferred as a starting point in development resulting in a moderate supercooling with the formation of moderate ice surface area.
Annealing
Annealing in certain instances can be utilized in a freeze drying process and usually serves two purposes, (1) crystallization of formulation component necessary to provide cake structure [37] and (2) increase the Tg’ of the amorphous by removal of crystalline component present with low Tg’ leading to a more efficient primary drying [16]. Additionally, annealing allows for redistribution of ice crystals resulting in an increase the pore size. For example, a higher drying temperature was achieved during lyophilization of a monoclonal antibody by incorporation of an annealing step essential for glycine crystallization [38]. Blue et. al. recently reported that addition of annealing step reduces the reconstitution time of an antibody formulation to 47 min as compared to a non-annealed sample (68 min) [20]. In contrast, annealing might also result in crystallization of an amorphous stabilizer thereby diminishing the stabilizing capacity [39]. Thus addition of an annealing step might have implication beyond efficient crystallization of bulking agent and should be considered on a case-by-case basis.
Primary Drying
Primary drying or ice sublimation removes the frozen water when the shelf temperature is raised and chamber pressure is reduced. Optimization of primary drying can yield large cycle time reduction and is dependant on formulation, shelf temperature, container and chamber pressure. Given the number of variables required to control the product temperature during primary drying, optimization of primary drying is considered a challenging task, with a significant economic impact, despite knowing the Tc and Tg’ of a given pharmaceutical formulation. Generally, the temperature at the ice-vapor interface should be several degree below Tc and/or Tg’ to avoid collapse [40]. On the other hand, higher efficiency and shorter cycle time can be obtained by operating at a temperature as close as possible to collapse temperature (recommended 2-5 oC below Tc). 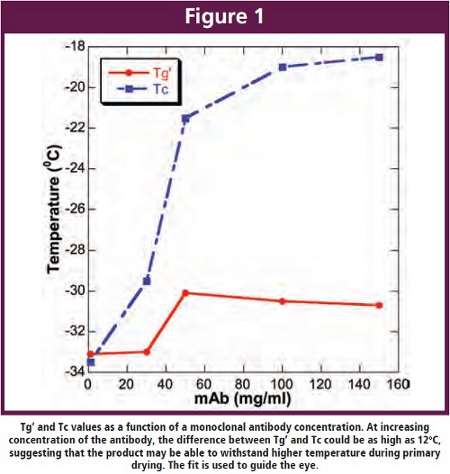 Additionally, it has been reported that operating at high temperature (scorch temperature) results in uniform distribution of temperature and bound water at the end of the cycle [41]. Therefore, an optimum temperature is one that provides a compromise between the freeze-drying time and safety. It should be noted that overloading of a freeze-dryer can occur at product temperature above -15oC [21]. Addition of a sugar or a polyol is usually recommended at a concentration sufficient (usually ≤ 10%) for stability purposes. Given the fact that addition of a saccharide increases the viscosity of the formulation matrix and reaction rate is inversely proportional to viscosity (from Stokes-Einstein relationship), freeze-drying of product at temperatures above Tg’ is worthwhile investigating [13,42]. Additionally, to avoid cake collapse, a bulking agent may be added as the collapse temperature of the bulking agent rich formulation is close to the eutectic temperature of the bulking agent (Teu for mannitol ~ -3oC while Tg’ of sucrose is ~ -34oC. Therefore at a high mannitol to sucrose ratio the Tc would be close to -5oC). The product temperature during drying is usually 5-40oC lower than the shelf temperature and depends on chamber pressure, heat transfer coefficient of the vials, the freeze dryer unit, formulation etc. As an approximate guideline, for every 5oC change of shelf temperature the product temperature changes by 1-20C [15,21]. The product temperature, as monitored using a thermal probe, is typically higher around the edges (edge effect) and colder in the interior because of radiative heat transfer from the chamber walls and the freeze-dryer’s door. Furthermore, to improve the rate of ice sublimation usually the drying is carried out at low pressure (50-200 mTorr). Very low pressure results in larger heterogeneity in heat transfer and might also cause contamination of products with pump oil or volatile stopper components [43,44]. Typically, a good starting point for development is 100 mTorr. Thus, an optimal shelf pressure allows for a high sublimation rate with homogenous heat transfer.
Additionally, it has been reported that operating at high temperature (scorch temperature) results in uniform distribution of temperature and bound water at the end of the cycle [41]. Therefore, an optimum temperature is one that provides a compromise between the freeze-drying time and safety. It should be noted that overloading of a freeze-dryer can occur at product temperature above -15oC [21]. Addition of a sugar or a polyol is usually recommended at a concentration sufficient (usually ≤ 10%) for stability purposes. Given the fact that addition of a saccharide increases the viscosity of the formulation matrix and reaction rate is inversely proportional to viscosity (from Stokes-Einstein relationship), freeze-drying of product at temperatures above Tg’ is worthwhile investigating [13,42]. Additionally, to avoid cake collapse, a bulking agent may be added as the collapse temperature of the bulking agent rich formulation is close to the eutectic temperature of the bulking agent (Teu for mannitol ~ -3oC while Tg’ of sucrose is ~ -34oC. Therefore at a high mannitol to sucrose ratio the Tc would be close to -5oC). The product temperature during drying is usually 5-40oC lower than the shelf temperature and depends on chamber pressure, heat transfer coefficient of the vials, the freeze dryer unit, formulation etc. As an approximate guideline, for every 5oC change of shelf temperature the product temperature changes by 1-20C [15,21]. The product temperature, as monitored using a thermal probe, is typically higher around the edges (edge effect) and colder in the interior because of radiative heat transfer from the chamber walls and the freeze-dryer’s door. Furthermore, to improve the rate of ice sublimation usually the drying is carried out at low pressure (50-200 mTorr). Very low pressure results in larger heterogeneity in heat transfer and might also cause contamination of products with pump oil or volatile stopper components [43,44]. Typically, a good starting point for development is 100 mTorr. Thus, an optimal shelf pressure allows for a high sublimation rate with homogenous heat transfer.
At the end point for primary drying, the product temperature increases to shelf temperature (transition temperature) signaling the end to primary drying. An additional soak time of at least 20% is recommended as drying in all vials is not finished at the same time. The end of primary drying can also be monitored using the Dew point sensor but the recommended method is the MTM/ Pressure rise method [15,21]. 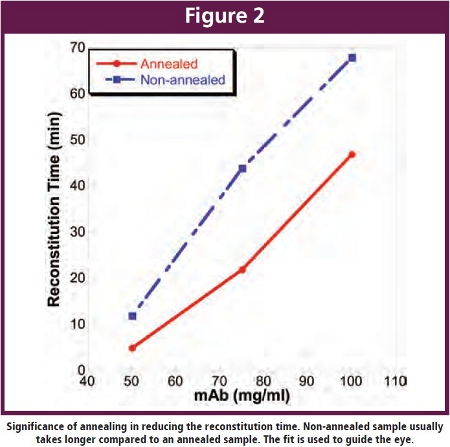 At high antibody concentrations, the total solute concentration usually exceeds 10% which in turn results in an increased resistance of dry layer to the flux of water vapor leading to prolonged drying times when operating at temperatures below Tg’. However, results from our experiments suggests that the difference between Tg’ and Tc increases as a function of increasing protein concentration (Figure 1) [20]. As a consequence, primary drying could be carried out at temperatures close to Tc with much shorter primary drying time. Also, it is well accepted that reconstitution time increases with increasing antibody concentration [12,10]. Results from our investigation has shown that a 2-fold decrease in reconstitution time can be achieved upon addition of annealing step during the freeze-drying process (Figure 2), although, the reconstitution times (~ 47 min) obtained under these conditions also puts constraints on the practical usage of the dried product [20]. Generally, high concentration antibody formulations can be obtained by lyophilizing a formulation comprising a lower antibody concentration and reconstituting to a higher antibody concentration [45]. The reconstitution time for a lyophilized cake prepared from 110 mg/ml bulk, for example, significantly dropped from 60 min to less than 5 min upon using the diluted bulk [46].
At high antibody concentrations, the total solute concentration usually exceeds 10% which in turn results in an increased resistance of dry layer to the flux of water vapor leading to prolonged drying times when operating at temperatures below Tg’. However, results from our experiments suggests that the difference between Tg’ and Tc increases as a function of increasing protein concentration (Figure 1) [20]. As a consequence, primary drying could be carried out at temperatures close to Tc with much shorter primary drying time. Also, it is well accepted that reconstitution time increases with increasing antibody concentration [12,10]. Results from our investigation has shown that a 2-fold decrease in reconstitution time can be achieved upon addition of annealing step during the freeze-drying process (Figure 2), although, the reconstitution times (~ 47 min) obtained under these conditions also puts constraints on the practical usage of the dried product [20]. Generally, high concentration antibody formulations can be obtained by lyophilizing a formulation comprising a lower antibody concentration and reconstituting to a higher antibody concentration [45]. The reconstitution time for a lyophilized cake prepared from 110 mg/ml bulk, for example, significantly dropped from 60 min to less than 5 min upon using the diluted bulk [46].
Secondary Drying
Secondary drying, the last stage of freeze-drying, removes the water from solute phase by desorption. Secondary drying is usually achieved at a slow ramp rate to avoid collapse of amorphous product (~ 0.1oC/min) while a higher ramp rate might be used for crystalline product (~ 0.5oC/min). Also, a recommended protocol is drying of product at high temperature for a short period of time rather than at low temperatures for longer times. This is attributed to the fact that water desorption rate decreases dramatically with time. An additional advantage of a short secondary drying process is the manufacturing benefit of the cabinet turnaround time. Desorption rate is sensitive to product temperature and does not depend on chamber pressure at pressure less than 200 mTorr. Secondary drying also depends on the solute concentration with higher solute concentration (>10%) resulting in longer drying times at a given temperature. It is well known that secondary drying facilitates achievement of the optimal residual moisture content (recommended ≤1%) necessary for desired storage stability of the product. Although, increasing the moisture content up to 5%, for certain antibody formulations might not alter the stability to a significant extent [20, 47].  Moisture content, in certain specific cases, might also impact the reconstitution time [20]. To ensure acceptable long-term stability, usually Tg (glass transition temperature) should be quantified as a function of moisture content [16]. At low levels, addition of every percent of moisture might lower the Tg by 100C or more [28]. Other factors might also play a significant role in storage stability. The Tg of antibody formulation containing sucrose, for example, was lower than that containing trehalose however greater stability was observed in the presence of sucrose probably due to the lower mobility in the sucrose formulation [48]. Therefore, we suggest that the exact conditions for stability should be empirically determined on a case-by-case basis.
Moisture content, in certain specific cases, might also impact the reconstitution time [20]. To ensure acceptable long-term stability, usually Tg (glass transition temperature) should be quantified as a function of moisture content [16]. At low levels, addition of every percent of moisture might lower the Tg by 100C or more [28]. Other factors might also play a significant role in storage stability. The Tg of antibody formulation containing sucrose, for example, was lower than that containing trehalose however greater stability was observed in the presence of sucrose probably due to the lower mobility in the sucrose formulation [48]. Therefore, we suggest that the exact conditions for stability should be empirically determined on a case-by-case basis.
Conclusion
Attainment of a stable high concentration antibody formulation with specific delivery device could be achieved through the use of proper antibody concentration and excipients, container/ closure, diluent and lyophilization cycle (Figure 3). Development of a successful lyophilized product can be a time- and energyconsuming process. In general, high concentration formulations are usually more resistant to both freezing and lyophilizationinduced antibody denaturation [28]. Given the complexity of lyophilization process and formulation design, attainment of a high concentration lyophilized formulation with a fast reconstitution time requires optimization of individual process parameters and proper understanding of the different freezing and drying stresses. This review provided an insight into the different stresses, their impact and approaches to mitigate them for achieving the highest drug quality at the least cost. Such efforts will ultimately lead the antibody-therapy to move into the “next-generation” with greater patient compliance.
References
1. Kohler, G, Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256: 495-497 (1975).
2. Reichert, JM, Dewitz, MC. Anti-infective monoclonal antibodies: perils and promises of development. Nature Rev. Drug Discov. 5: 191-195 (2006).
3. Lefranc, MP, Giudicelli, V, Kaas, Q, Duprat, E, Jabado-Michaloud, J, Scaviner, D, Ginestoux, C, Clement O, Chaume, D, Lefranc, G. IMGT, the international ImmunoGenetics information system. Nucleic Acids Res. 33: D593-D597 (2005).
4. Reichert, JM, Rosensweig, CJ, Faden, LB, Dewitz, MC. Monoclonal antibody successes in the clinic. Nature Biotechnol. 23: 1073-1078 (2005).
5. Glatz, CE. Modeling of aggregation-precipitation-phenomena. In Aherns, TJ, Manning, MC, eds. Stability of protein pharmaceuticals, 1st edition, New York, Plenum Press. P 135-166 (1992).
6. Treuheit, MJ, Kosky, AA, Brems, DN. Inverse relationship of protein concentration and aggregation. Pharm. Res. 19(4): 511-516 (2002).
7. Minton, AP. Influence of macromolecular crowding upon the stability and state of association of proteins: predictions and observations. J. Pharm. Sci. 94:1668-1675 (2005).
8. Minton, AP. The influence of macromolecular crowding and macromolecular confinement on biochemical reactions in physiological media. J. Biol. Chem. 276: 10577-10580 (2001).
9. Liu, J, Nguyen, MD, Andya, JD, Shire, SJ. Reversible self-association increases the viscosity of a concentrated monoclonal antibody in aqueous solution.
10. Shire, SJ, Shahrokh, Z, Liu, J. Challenges in the development of High Protein Concentration Formulations. J. Pharm. Sci. 93: 1390-1402 (2004).
11. Atkinson, EM: Klum, W. Formulations strategies for biopharmaceuticals-ensuring success to market. IDrugs 4: 557-560 (2001).
12. Harris, RJ. Heterogeneity of recombinant antibodies: linking structure to function. Dev. Biol. (Basel) 122: 117-127 (2005).
13. Privalov, PL. Cold Denaturation of proteins. Crit. Rev. Biochem. Mol. Biol. 25: 281-305 (1990).
14. Carpenter, JF, Prestrelski, SJ, Arakawa, T. Separation of freezing- and drying-induced denaturation of lyophilized proteins using stress-specific stabilization. I. Enzyme activity and calorimetric studies. Arch. Biochem. Biophys. 303: 456-464 (1993).
15. Pikal, MJ Lyophilization. In Swarbrick, J, Boylan, J (eds.) Encyclopedia of Pharmaceutical Technology, Marcel Dekker, New York, 1299-1326 (2002).
16. Carpenter, JF, Pikal, MJ, Chang, BS, Randolph, TW. Rational Design of Stable Lyophilized Protein Formulations: Some Practical Advice. Pharm Res. 14: 969-975 (1997).
17. Harris, JH, Shire, SJ, Winter, C. Commercial Manufacturing Scale Formulation and Analytical Characterization of Therapeutic Recombinant Antibodies. Drug Dev. Res. Vol. 61: 137-154 (2004).
18. Sarciaux, JM, Mansour, S, Hageman, MJ, Nail, SL. Effects of Buffer conditions on Aggregation of Bovine IgG during Freeze-Drying. J. Pharm. Sci. 12: 1354-1361 (1999).
19. Chang, BS, Kendrick, BS, Carpenter, JF. Surface- Induced Denaturation of Proteins during Freezing and its Inhibition by Surfactants. J. Pharm. Sci. 12:1325-1330 (1996).
20. Blue, J, Yoder, H. Successful lyophilization development of protein therapeutics. Am. Pharm. Rev. 40-44 (2009).
21. Tang, XC, Pikal, MJ. Design of Freeze-Drying Processes for Pharmaceuticals: Practical Advice. Pharm. Res. 2: 191-200 (2004).
22. Pikal, MJ. Freeze-Drying of proteins. Part I: process design. BioPharm 3: 18-28 (1990).
23. Pikal, MJ. Freeze-Drying of proteins. Part II: formulation selection. BioPharm 3: 26-30 (1990).
24. Lueckel, B, Bodmer, D, Helk, B, Leuenberger, H. Formulations of sugars with amino acids or mannitol-influence of concentration ratio on the properties of the freeze-concentrate and the lyophilizate. Pharm. Dev. Technol. 3: 325-336 (1998).
25. Williams, NA, Lee, Y, Polli, GP, Jennings, TA. The effects of cooling rate on solid phase transitions and associated vial breakage occurring in frozen mannitol solutions. J. Parenter. Sci. Technol. 40: 135-141 (1986).
26. Searles, JA, Carpenter, JF, Randolph, TW. Annealing to optimize the primary drying rate, reduce freezing-induced drying rate heterogeneity, and determine Tg’ in pharmaceutical lyophilization. J. Pharm. Sci. 90: 872-887 (2001).
27. Pikal, MJ, Shah, S, Roy, ML, Putman, R. The secondary drying stage of freeze drying: drying kinetics as a function of temperature and chamber pressure. Int. J. Pharm. 60: 203-217 (1990).
28. Wang, W. Lyophilization and development of solid protein pharmaceuticals. Int. J. Pharm. 203: 1-60 (2000).
29. Franks, F. Freeze-drying: from empiricism to predictability. Cryo-Letters 11: 93-110 (1990).
30. Middaugh, CR, Gerber-Jenson, B, Hurvitz, A, Paluszek, A, Scheffel, C, Litman, G., Physicochemical characterization of six monoclonal cryoimmunoglobulins: possible basis for cold-dependent insolubility. Proc. Natl. Acad. Sci. U.S.A. 75: 3440-3444 (1978).
31. Dill, KA, Alonso, DOV, Hutchinson, K. Thermal stabilities of globular proteins. Biochemistry, 28: 5439-5449 (1990).
32. Jaenicke, R. Protein structure and function at low temperatures. Philos. Trans. R. Soc. Lond. B Biol. Sci. 326: 535-551 (1990).
33. Franks, F. Solid aqueous solutions. Pure Appl. Chem. 65, 2527-2537 (1993).
34. Chang, BS, Randall, CS. Use of subambient thermal analysis to optimize protein lyophilization. Cryobiology, 29: 632-656 (1992).
35. Hsu, CC, Walsh, AJ, Nguyen, HM, Overcashier, DE, Koning-Bastiaan, H, Bailey, RC, Nail, SL. Design and application of a low-temperature Peltier-cooling microscope stage. J. Pharm. Sci. 85: 70-74 (1996).
36. Kim, AI, Akers, MJ, Nail, SL. The physical state of mannitol after freeze-drying:effect of mannitol concentration, freezing rate, and noncrystallizing cosolute. J. Pharm. Sci. 87: 931-935 (1998).
37. Getlin, LA. Kinetics of Phase transitions in a frozen solution. Dev. Biol. Stand. 74: 93-104 (1996).
38. Ma, X, Wang, DQ, Bouffard, R, MacKenzie, A. Characterization of protein formulation for freeze-drying cycle development. Pharm. Sci. 1 (Suppl.) S543 (1998).
39. Lueckel, B, Helk, B, Bodmer, D, Leuenberger, H. Effects of formulation and process variables on the aggregation of freeze-dried interleukin-6 (IL-6) after lyophilization and on storage. Pahrm. Dev. Technol. 3: 337- 346 (1998b).
40. Mackenzie, AP. Basic Principles of Freeze-Drying for Pharmaceuticals. Bull. Parenter. Drug Assoc. 20: 101-130 (1966).
41. Sheehan, P, Liapis, AI. Modeling of the primary and secondary drying stages of the freeze drying of pharmaceutical products in vials: numerical results obtained from the solution of a dynamic and spatially multi-dimensional lyophilization model for different operational policies. Biotechnol. Bioeng. 60: 712-728 (1998).
42. Kramers, HA. Brownian motion in a field of force and diffusion model of chemical reactions. Physica 7: 284-304 (1940).
43. Pikal MJ, Roy, ML, Shah, S. Mass and heat transfer in vial freeze-drying of pharmaceuticals: role of the vial. J. Pharm. Sci. 73: 1224-1237 (1984).
44. Pikal, MJ, Lang, JE. Rubber closures as a source of haze in freeze dried parenterals: test methodology for closure evaluation. J. Parenter. Drug Assoc. 32: 162-173 (1978).
45. Andya JD, Gwee, SC, Liu, J, Shen, Y. U.S. Patent 6,267,958 (2006).
46. Breen, ED, Constantino, HR, Hsu, CC, Shire, SJ. Affects of bulk concentration on reconstitution of lyophilized protein formulations. Pharm. Sci. 1 (Suppl.), S540 (1998).
47. Kataka, M, Robillard, P, Brown, J, Tolman, G. Effects of moisture and lyophilization cycle on aggregation of a monoclonal antibody product. Pharm. Sci. 1 (Suppl.) S543 (1998).
48. Duddu, SP, Zhang, G, Dal Monte, PR. The relationship between protein aggregation and molecular mobility below the glass transition temperature of lyophilized formulations containing a monoclonal antibody. Pharm. Res. 14: 596-600 (1997).
Akhilesh Bhambhani, PhD is a Senior Research Chemist at the Merck & Co., Inc., (West Point, PA). He earned his B.S. in Chemical Engineering in 2001 from Ujjain Engineering College, India and his PhD in Biological Chemistry in 2006 from University of Connecticut (Storrs, CT). Prior to joining Merck in 2009, Akhilesh worked as a postdoctoral researcher at The University of Kansas (Lawrence, KS) and later as a Development Scientist I at Amylin Pharmaceuticals (San Diego, CA) developing various liquid and lyophilized products for biologics including high concentration antibody formulation and formulations of peptides, proteins, bacteria and vaccines. In his current role, he is primarily focused on development of a platform lyophilized formulation for therapeutic drug candidates at high concentration, lyophilization cycle and process development. His expertise in this area was recently recognized by Merck with an “award of excellence”. He can be contacted by telephone: (215) 652- 7466 or by email: [email protected].
Jeffrey T. Blue received his BS degree in Biology with a concentration in genetics and biochemistry from Carnegie Mellon University in Pittsburgh followed by completing a MS degree in Biochemistry from Lehigh University. He has been at Merck and Co. Inc for 15 years and is the current leader of both the preformulation and formulation group for all Vaccines and Biologics. He has played major roles in the development of lyophilization for three marketed vaccine products and is currently developing lyophilization platforms for biologics. His expertise in this area was recently recognized by Merck with a special achievement award for his contribution in developing a lyophilization cycle and process to successfully manufacture Zostavax®.