Abstract
Over the last decade, a large number of publications and pharmaceutical conferences dealt with drug delivery strategies for an increasing number of poorly water soluble and lipophilic drugs. To deliver these appropriately, typically it requires application of non-standard formulation technologies (apart from salt formation and micronization), e.g. lipid/surfactant based SEDDS/SMEDDS (self-emulsifying/self-microemulsifying) or solid dispersion formulation approaches. Most recently many molecules cannot be well delivered even when they are formulated with these tools due to low solubility in lipidic and polymeric excipients.
Nanoparticulate formulation approaches could be applied instead as alternative. As in the case of solid dispersions, successful penetration of these generally rather difficult to develop (i.e. due to meta-stability) approaches into commercial drug product development has taken, also for nanoparticulate based formulations, many years.
The article tries to give a helicopter perspective overview about nanoparticulate and solid dispersions formulation technologies, underestimated similarities between both technologies. Screening options for feasibility studies, technical challenges regarding stabilization, further processing and up-scaling aspects are also discussed. A few case examples illustrate the landscape. Further details can also be found in the references at the end of this article.
Concepts Behind Nanoparticulates and Solid Dispersion Approaches
The enormous interest in enhancing drug delivery strategies due to an increasing number of poorly soluble drug candidates has been widely discussed and is reflected in literature and presentation at drug delivery conferences [1, 2, 3, 4, 5] . The reasons why solid dispersions and nanoparticulate based formulations had been mainly in the focus only of academia but not of major parts of pharmaceutical industry are manifold:
- Meta-stable nature of the systems (e.g. physical instability/ re-crystallization risks of solid solution/dispersions with the potential of deteriorating drug release and bioavailability)
- Lack of appropriate manufacturing and analytical characterization tools available in-house (e.g. solvent based technologies for preparing solid dispersions with the need to have expensive solvent recovery systems [6])
- Lack of experience and trained personnel to apply these systems effectively in drug product development, to prepare these systems in-house and screen them analytically (e.g. more sophisticated DSC tools like modulated DSC, particle size and zeta potential analysis for nanoparticles, polymorphic characterization of nanoparticles)
- Proprietary technologies owned by few specialist technology companies (i.e. in the field of making nanoparticles or melt extruded solid dispersions) with the concern to infringe their technology patents
- Lack of sufficient senior management support to invest in these more risky technologies
The typical way how these gaps could be overcome is that in the starting phase a few companies take the huge risk to bring such a technology to the market, i.e. due to an urgent drug delivery and patient need, i.e. improve bioavailability by dramatic particle size reduction into the nanometer range.
The Fenofibrate Case – Improvements with Nanoparticles
A good example is the case with the lipid lowering drug Fenofibrate: Abbott had brought Lipidil® with 100 mg unmicronized drug as a capsule on the market. This formulation was replaced in the US by Tricor® Capsules Micronized (67, 134, 200, 267 mg) and then finally by a nanocrystalline formulation, marketed as Tricor® NanoCrystal (48, 145 mg) tablets. With the dramatic drop in particle size from the μm ranges down to the nanometer scale, an enhancement in bioavailability was achieved which allowed a significant dose reduction and the relief not to take the drug in each case together with food to achieve similar blood levels.
Another example for the benefit of nanoparticulates could be a significant food reduction as has been published by J. Jinno et al. [7]: Cilostazol, a BCS class II compound, shows a positive food effect which can be dramatically reduced in a beagle dog study when the drug is nanosized using nanomilling technology.
The primary bioavailability mechanism for nanoparticulate formulations is a leverage in dissolution rate due to a large increase of the surface area A at smaller particle sizes as shown by the Noyes- Whitney equation (see equation 1):
Equation 1:
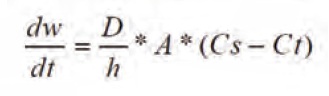
Dw/dt=Dissolution rate
D=Diffusion coefficient
h=Thickness of diffusion layer
A=Interfacial surface are
Cs=Saturation solubility
Ct=Solubility at t
In addition to a surface area increase, the saturation solubility of the drug compound is leveraged due to a higher intrinsic dissolution (“vapor”) pressure for smaller particles in the lower submicron range due to much higher particle curvatures (smaller radius of curvature) – this relationship is mathematically described by the Freundlich-Ostwald equation and has been estimated to contribute to approximately 10 – 50 % of saturation solubility increases [8].
In case of solid dispersion formulations, the dissolution rate can also be dramatically increased (similar to nanoparticulate drugs), if the compound is either in its pure amorphous form - assuming sufficient wettability of the drug surface by the dissolution mediumor amorphously embedded in a suitable carrier matrix which can be typically a pharmaceutical polymer or a polymer plus a surfactant or another carrier like urea or a sugar alcohol (e.g. Mannitol): In this case, no crystal lattice energy needs to be thermodynamically overcome which fosters then fast dissolution.
But it has also been recently reported that nanoparticulate structures were observed and measured when sold dispersion particles started to dissolve, i.e. Bikaris et al. found small nanometer particles when Felodipine/PVP solid dispersions with 10 – 20 % drug load were dissolved in pH 6.5 phosphate buffer solutions (with 2 % Polysorbate 20, 100 rpm paddle) [9]. So it seems as if there are ultrafine nanometer range particles available once the solid dispersion starts to dissolve – a dramatic increase in surface area beside much higher intrinsic saturation solubility due to having no crystal lattice energies that needs to be overcome in solution.
Overcoming Drug Precipitation
Another extremely important aspect of developing the right formulations for a poorly soluble drug from the beginning is the problem that pH-sensitive molecules like weak bases or weak acids can undergo dramatic changes in solubility during their gastrointestinal passage, i.e. solubility in the gastric juice might be sufficiently high to dissolve the weak base or a corresponding salt. But with the entrance of the dissolved drug into gastric regions of higher pH, e.g. in the small intestine, solubility might drop dramatically associated with a precipitation of the corresponding free drug base into particles of increasing sizes which have then a low probability of getting re-dissolved and absorbed in later compartments of the gut. A similar situation could happen with ingestion of a meal:
- D. Hörter and J. Dressman reported about postprandial pH increases up to 5 circa 0.5 – 1 hour after meal intake – this could then already be the trigger for converting a soluble salt species into its insoluble corresponding precipitate.
- The following figure 1 depicts a dramatic particle size increase of a poorly soluble weak base given as salt from the first measurement after 10 minutes up to one hour in a pH 6.8 buffer solution and FaSSIF and FeSSIF dissolution media: While size ranges of a few 10 μm dominated after 10 minutes, particle growth up to large 100 μm particles occurred within the first hour.
Proper characterization of the precipitates (i.e. particle size ranges, type of polymorph formed) is important to make an assessment about opportunities of re-dissolution and later absorption. Formation of a precipitated very insoluble hydrate for instance would give a low chance that this might actually occur. In some cases, occurrence of double-peaks in the blood plasma levels could give a hint that the precipitated drug has got a 2nd chance for absorption (if no entero-hepatic circulation would be responsible for this phenomenon). Suitable in-vitro dissolution systems to investigate drug precipitation have been described in literature, e.g. [10, 11]. More elaborated models combine a precipitation model with a Caco-2 cell model: The dissolution fluid at pH 6.8 is pumped into the donor compartment where the drug can then penetrate through a Caco-2 cell layer. The drug concentration in the acceptor compartment is the measured corresponding to fractions of absorbed drug [12].
 Since the bioavailability enhancing mechanism of nanoparticulate approaches is fast dissolution, per se, there is no principle integrated into these systems which would efficiently avoid drug precipitation. On the opposite side, as in the case of solid dispersion formulations and SEDDS/SMEDDS formulations, the soluble drug is kept for a certain time length in an oversaturated solution status stabilized by the polymeric or surfactant rich microenvironment. In a 20% (w/w) solid dispersion eighty percent of the polymerare present to potentially stabilize the oversaturated system which would not be the case for nanosuspensions with typically much lower polymer concentrations. Oversaturation allows poorly water soluble drugs that are rapidly absorbed to permeate through the intestinal cellular membranes within a certain timeframe which can lead to dramatic increases in bioavailability in many cases.
Since the bioavailability enhancing mechanism of nanoparticulate approaches is fast dissolution, per se, there is no principle integrated into these systems which would efficiently avoid drug precipitation. On the opposite side, as in the case of solid dispersion formulations and SEDDS/SMEDDS formulations, the soluble drug is kept for a certain time length in an oversaturated solution status stabilized by the polymeric or surfactant rich microenvironment. In a 20% (w/w) solid dispersion eighty percent of the polymerare present to potentially stabilize the oversaturated system which would not be the case for nanosuspensions with typically much lower polymer concentrations. Oversaturation allows poorly water soluble drugs that are rapidly absorbed to permeate through the intestinal cellular membranes within a certain timeframe which can lead to dramatic increases in bioavailability in many cases.
Nanoparticulate Formulations
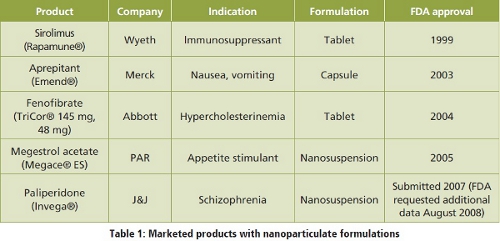
The following table 1 shows currently marketed products based on nanoparticlulate formulations:
Manufacturing Technologies
Nanoparticles can generally be prepared in two different ways, either by
- Top-down technologies: wet-milling/ball-milling or high pressure homogenization. The crystal structure is mechanically affected by mechanical energies which lead to crystal destruction. Partial or total amorphisation or polymorph conversion can occur. Nanoparticles are stabilized by surfactants (electrostatic) and/or polymers (steric).
- Bottom-up technologies: in these cases the drug typically is dissolved in a solvent together with a stabilizer. The drug solution is precipitated e.g. through a nozzle into an anti-solvent. Formed nanoparticles are stabilized by surfactants (electrostatic) and/or polymers (steric)
 Typically initial nanoparticulate products are in aqueous solution. Further processing into solid oral dosage forms is possible i.e. by freeze- or spray-drying or spray-granulation together with suitable carrier excipients (Lactose, Mannitol, Microcrystalline Cellulose etc.). Figure 2 shows schematically the manufacturing flow for a bottom-up process:
Typically initial nanoparticulate products are in aqueous solution. Further processing into solid oral dosage forms is possible i.e. by freeze- or spray-drying or spray-granulation together with suitable carrier excipients (Lactose, Mannitol, Microcrystalline Cellulose etc.). Figure 2 shows schematically the manufacturing flow for a bottom-up process:
Wet Ball Milling
Among the top-down technologies especially the ball-milling process has led to successful launches of the above mentioned marketed products. The proprietary technology is owned by one company which had developed it already circa 1990. Their applications have been widely described in patents and literature [e.g. 13-15]. In the ball milling process typically the drug is already suspended in its micronized form in an aqueous medium to facilitate the nanomilling process. The milling chamber contains the raw suspension together with a grinding medium, i.e. zirconium oxide, polystyrene or glass beads of less than 1 mm. A motor driven mechanical agitator shaft bears the impeller and provides high-shear agitation up to several thousand rpm. The fluid is pumped through a media separator screen and back again into the milling chamber. Mill and outside re-circulation vessel are jacketed to control the temperature sufficiently (e.g. inlet temperature 5-10°C) .Milling chamber sizes from 300 ml up to 60-L allow for up-scaling of the process from lab into production scale. Figure 3 shows a schematic drawing of a ball mill.
High-pressure Homogenization
High-pressure homogenization has been evaluated in many publications by R.H. Müller et al (e.g. a good overview is given in [16]). A macro-suspension containing an aqueous surfactant for instance is pressures through a small gap of 20 – 30 μm with a piston. The streaming velocity of the fluid is dramatically increased by pushing the material through the gap.  Since the static pressure drops behind the gap, water starts to boil. Implosion of the resulting water gas bubbles creates very strong cavitational forces that lead to mechanical destruction of the crystals. The technology has also been used commercially by several companies and led to marketed products (i.e. applied for Fenofibrate).
Since the static pressure drops behind the gap, water starts to boil. Implosion of the resulting water gas bubbles creates very strong cavitational forces that lead to mechanical destruction of the crystals. The technology has also been used commercially by several companies and led to marketed products (i.e. applied for Fenofibrate).
Stabilization of Nanoparticles
One of the very important aspects of nanoparticles is that they typically have the tendency to re-agglomerate due to increasing importance of attractive inter-particulate forces (van der Waals forces) in the nanometer range. Another important phenomenon that is described as `Ostwald ripening` is based on an imbalance between the higher saturation concentration in the vicinity of very small particles (higher diffusion/”vapor” pressure due to higher curvature!) and lower concentrations close to larger particles. Smaller particle fractions vanish over time due to higher solubility while larger particles fractions start to grow.
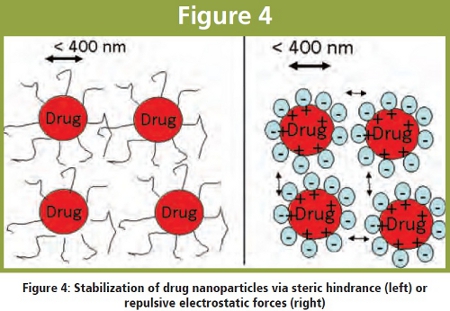
To overcome these phenomena, nanoparticles can be separated by fostering repulsive forces among them (electrostatic stabilization e.g. by adding ionic surfactants or charged polymers) or keeping them apart via steric hindrance by adsorbing polymeric stabilizers such as Hydroxypropyl methyl cellulose (HPMC), Hydroxypropyl cellulose (HPC), Povidone (PVK K 30) and copolymers like Pluronic F68 or F127 (see figure 4). Chain lengths of these molecules should be not too long to slow down dissolution.
 Recently another stability phenomenon has been described as `gel relaxation` by Z. Deng et al. [17]: Shortly after wet-milling particle agglomeration/cluster formation has been observed with dramatic morphology changes under the SEM: With the particle growth up to a maximum a significant viscosity increase has been measured which dropped dramatically when the nanosuspension was standing for more than 24 hours (gel relaxation). By modifying milling conditions (extended milling time, increased concentration of surfactants etc.) the extent of this phenomenon could be reduced.
Recently another stability phenomenon has been described as `gel relaxation` by Z. Deng et al. [17]: Shortly after wet-milling particle agglomeration/cluster formation has been observed with dramatic morphology changes under the SEM: With the particle growth up to a maximum a significant viscosity increase has been measured which dropped dramatically when the nanosuspension was standing for more than 24 hours (gel relaxation). By modifying milling conditions (extended milling time, increased concentration of surfactants etc.) the extent of this phenomenon could be reduced.
Adsorption of positively or negatively charged particles at the charged drug surface leads to a electrical double-layer and a specific electrical charge of drug nanoparticle which is decreasing with increasing distance from nanoparticle surface. The potential at the slipping/boundary plane is expressed as `zeta potential` measured in mV units. 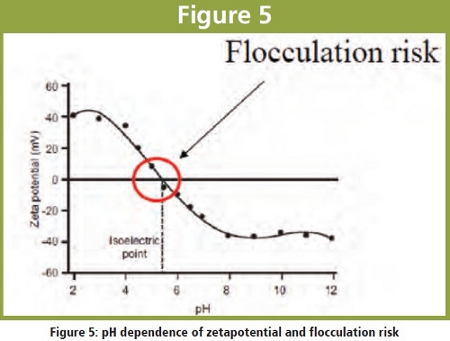 The zeta potential should be typically at least 30 mV. pH dependency of the zeta potential is shown in figure 5: At 0 mV flocculation risk becomes very high (absence of interparticular repulsive forces !).
The zeta potential should be typically at least 30 mV. pH dependency of the zeta potential is shown in figure 5: At 0 mV flocculation risk becomes very high (absence of interparticular repulsive forces !).
Screening of Nanoparticulate Formulations
How to efficiently design stable nanoparticulate formulations, for instance with polymeric or ionic surfactant stabilizers, is currently more a rather trial and error based screening than a strict science based approach.
L. Lindfors et al. described rational stabilization of amorphous Felodipine nanoparticles with middle-chain triglycerides (MCT) prepared by a solvent quench method. Ostwald ripening rate was slower than predicted from LSW (Liftshitz, Slyozow,Wagner) model equation when MCT was present. Ripening was only efficient in other molecule cases, when MCT was completely miscible with the drug. Interaction can be calculated by a Bragg-Williams chi parameter c : Particle growth inhibition was efficient when c was < 2 [18]. Miscibility of the drug with the inhibitor was also monitored via 1H-NMR spectroscopy.
J. Lee evaluated the combinations of 5 common polymer stabilizers, 2 low molecular weight surfactants and 11 insoluble drugs with keeping processing and characterization conditions the same [19]: 2 ml vials with 500 μm polystyrene beads were used and milling was performed on a high-speed shaker. Correlation attempts of the drug substance and polymer surface energies (determined via static contact angle measurements) with the milling results (particle sizes) are not really straightforward; nevertheless in several instances similar surface energies seem to be favorable. Poor stabilization results gained with PEG were interpreted as lack of hydrophobic units. As a rule of thumb:
- drugs of high molecular weight,
- low solubility,
- high melting points,
- a surface energy similar to that of the polymers can be successfully transformed to nanoparticles of unimodal particle size distribution.
- Adding surfactants results in an additional size reduction in certain polymer/drug pairs,
- both anionic and cationic surfactants produce similar size reductions in a polymer/drug pair.
B. van Eerdenbrugh et al. have recently published a nanosuspension scaling-down production study by milling compounds in glass vials in a ball mill which allows for screening stable nanosuspension formulations with very low drug substance amounts of circa 10 mg and less (1 mg) per experiment [20]. Analytical characterization of these nanosuspensions (particle size by laser light scattering, SEM, DSC and solid state characterization by XRPD) allowed for efficient formulation screening.
Transformation into Solid Products
Solid oral dosage forms represent from a market and patient convenience standpoint the majority of all drug delivery forms. Therefore, one very important aspect for nanoparticulate formulations is how to transfer these into a solid oral product: i.e. granules that could be further compressed into a tablet which might be further film-coated etc. Technically this could be achieved by e.g. spray-drying, freeze-drying or spray granulation [21]. Key aspect of such solidified nanosuspensions is the maintenance of the re-dispersibility, hence generation of a nanoparticulate size distribution once the solid form comes into contact with gastric or intestinal fluids. Different carriers like Sucrose and Mannitol for freeze-drying, Mannitol, Microcrystalline Cellulose, Anhydrous Calcium Phosphate, colloidal Silicon Dioxide and a hydrophobically modified Inuline for spray-drying were evaluated for a set of nine poorly soluble drugs [22]. It has been found that drugs with more hydrophobic surface depict disintegration delays, which underlines the strong need for integrating appropriate carrier excipients: I.e. the hydrophobically modified Inuline showed superior performance due to surface active properties.
The following figure shows a Novartis compound that has been spray-dried by embedding the drug nanoparticles (ca. 320 nm) in PVP, Lactose or Mannitol as carrier excipients (drug load 47%).. Highest dissolution rates under non-sink conditions were achieved with PVP > Lactose > Mannitol.
Significant differences could be observed under the electron microscope: The PVP embedded particles showed the smoothest surfaces of the spray-dried particles.
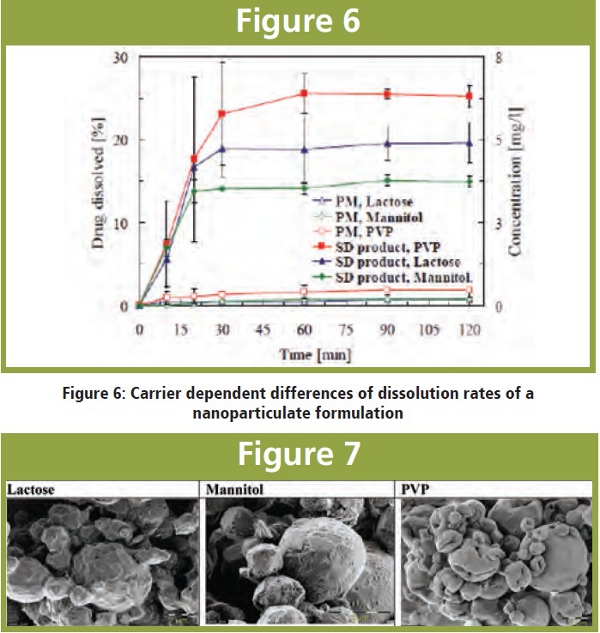
Figure 7: SEM images of spray-dried nanoparticulate formulations
Solid Dispersion Formulations
The drug solubility enhancing principle of solid dispersions is based ideally on an amorphous and molecular dispersion of a poorly soluble drug in a carrier matrix which allows to improve dissolution of the drug in water or in physiological media since no crystal lattice energy needs then to be overcome. Typically an oversaturated solution is generated, bearing the risk of fast recrystallization/precipitation.
Three different generations of solid dispersions can be differentiated [24]:
- 1st generation solid dispersions (1960s) using crystalline carriers like Fructose, Urea, Mannitol: They lead to an improvement of wettability, but require again energy to break up the crystalline structures of the carrier
- 2nd generation solid dispersions (1970s) using polymeric carriers like PVP, PVA, HPMC, HPC, Ethylcellulose, PEG: At that time systems were often heterogeneous mixtures of amorphous drug solutions in the polymer carrier and non-dissolved small microcrystalline particles due to only partial miscibility.
- 3rd generation solid dispersions (1990s until now): In these modern systems typically the drug is kept in an amorphous (or nanocrystalline) state, no crystal structures need to be broken up during dissolution. Inclusion of surface active substances and/ or carriers (Inulin, Lauroyl Macrogolglycerides or Glyceryl Behenate) improve precipitation robustness (see discussion above) and increase bioavailability of these solid dispersions.
Table 2 (Next Page) gives an overview about marketed solid dispersion formulations (see [6])
Manufacturing Technologies
Classical methods of preparing solid dispersion formulations are:
- the melting method (melt extrusion) and
- solvent evaporation techniques [4].
The melting processes require often high temperatures >> 100°C which may lead to thermal degradation of the drug substance. Melt extrusion has offered here an attractive, solvent-free solution to extrude thermoplastic materials (polymers) together with dissolved drugs homogeneously at much lower temperatures than the melting point of the drug and the softening temperature of the polymer [5].
A more detailed overview about solvent based manufacturing processes for solid dispersions is given in [6].
A melt extrusion processes is compared against a solvent coprecipitation based in [23]. Typically for melt extrusion processes drug substances are extruded together with a polymer at high temperatures > 100°C in a twin-extruder to dissolve the drug in the molten polymeric carrier matrix.  The mechanical shearforces applied during extrusion represent -apart from the high temperature- another factor that fosters dissolution of the poorly soluble compound in the polymer matrix. Twin-screw extruders comprise - apart from shearing elements - heating and cooling segments and allow for short residence times and reduced heating stress due to the continuous mass flow. Manufacturing processes are typically developed on a drug substance and carrier specific basis. Homogeneity of the extrudates should be carefully checked; application of confocal Raman spectroscopy has for instance been described as a tool by J. Breitenbach [25].
The mechanical shearforces applied during extrusion represent -apart from the high temperature- another factor that fosters dissolution of the poorly soluble compound in the polymer matrix. Twin-screw extruders comprise - apart from shearing elements - heating and cooling segments and allow for short residence times and reduced heating stress due to the continuous mass flow. Manufacturing processes are typically developed on a drug substance and carrier specific basis. Homogeneity of the extrudates should be carefully checked; application of confocal Raman spectroscopy has for instance been described as a tool by J. Breitenbach [25].
Physical Stabilization Principles of Solid Dispersions
Solid dispersions can principally be stabilized by
- Thermodynamic stabilization: In this case, the drug concentration is kept below the saturation solubility e.g. in the polymeric carrier matrix. Testing of drug candidates in dimeric corresponding components has been shown to be predictive regarding saturation solubility [25].
- Kinetical stabilization: In case of less soluble drugs in the polymer, the viscosity of the resulting solid dispersion – typically expressed via the glass transition temperature Tg - should be sufficiently high (recommend 40 – 50 K above the intended storage temperature) [26].
Physical stability predication of solid dispersion formulations is not an easy task and normally real time stability data are used to generate relevant information for regulatory purposes. Angell plots which monitor the characteristic relaxation time τ (relaxation time of the glass transition signal) via modulated DSC over the ratio Tg/T (glass transition temperature divided by the storage temperature) have been modified to make re-crystallization predictions. Lowering of the storage temperature leads to an increase of the Tg/T ratio and leverages the relaxation time τ - the increased viscosity of the solid dispersion helps to avoid drug re-crystallization in the amorphous matrix.
Screening of Solid Dispersions
Solid dispersion formulations can be analyzed with different analytical techniques regarding
- In-vitro performance in water, physiological media (SGF, FaSSIF, FeSSIF),
- physical stability: XRPD (absence of crystallinity), DSC/ mDSC (monitoring of Tg), SEM, GC and Karl Fischer (content of residual solvents and water acting as destabilizing plasticizers lowering Tg)
With the Tg determination, solubility of the drug compound in the polymer can be monitored: In case of good solubility only a single Tg signal becomes visible. Each phase separation (i.e. separation of the amorphous drug phase from the polymer phase) can lead to separation of individual Tg signals [6].
Solid Dispersions – Carrier Embedded Nanoparticles?
As discussed above, it has been found by D. Bikirias et al [9], that solid dispersion particles at nanometer can be formed by dissolving a Felodipin/PVP solid dispersion in a pH 6.5 phosphate buffer solution with 2 % polysorbate. An advantage of solid dispersions over pure dried nanosuspensions might be a more efficient stabilization of the oversaturation of the dissolved particles due to much higher polymer concentrations. Typically solid dispersions are formulated as tablets which should normally disintegrate slower when compared to a conventional immediate release tablet with just a few percents of polymer as a binder. Many solid dispersions have high polymer concentrations up to 80 – 90 %.
In an internal in-vivo beagle dog study, a comparison has been made between a larger monolithic tablet with a solid dispersion of a poorly soluble drug (no pH dependent solubility) compound (drug load 20% w/w) against a multi-particulate solid dispersion mini-tablet formulation which was in addition coated with a gastric resistant polymer to prevent a too early drug precipitation in the stomach.
The following figure shows that the monolithic tablet gave slightly higher drug exposure (bioavailability) compared to the small multi-particulates with larger surface area:
Obviously the slower polymer erosion of the monolithic solid dispersion with reduced free accessible surface could be somewhat beneficial with respect to prevent a rapid drug precipitation out of the oversaturated drug concentration state. Similar observations were made for glassy solid dispersion melt-extrudates which protect against drug precipitation for a certain while.
Summary and Conclusions
Introduction of nanoparticulate and solid dispersion formulations into routine industrial drug product development practice has taken a few decades until first products showed up in the drug markets. Nowadays, these special drug delivery systems are becoming more and more important with the increasing number of poorly soluble drugs in early development stages. 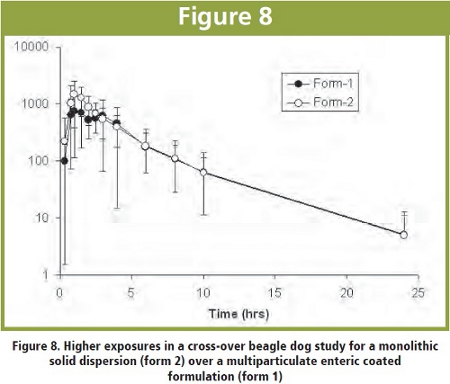 Especially nanosuspension based formulations have gained strong interest now in cases where drugs depict only marginal solubility in lipidic and surfactant excipients and polymers, making development of a SEDDS/SMEDDS or solid dispersion nearly impossible.
Especially nanosuspension based formulations have gained strong interest now in cases where drugs depict only marginal solubility in lipidic and surfactant excipients and polymers, making development of a SEDDS/SMEDDS or solid dispersion nearly impossible.
Typically many “brick dust” molecules with high melting points > 150 – 200°C could be interesting candidates for this technology, whereas “grease ball” drugs should be better formulatable with a lipidic system.
Nanoparticulate and solid dispersions seem to have some similarities since drug particles in the nanometer range play a role during dissolution in both cases.
Precipitation robustness is another extremely important aspects – further research work is required to optimally develop these enhanced formulations with improved performance under real in-vivo conditions.
References
1. C. Timpe, Strategies for Formulation Development of Poorly Water-Soluble Drug Candidates – a Recent Perspective, Am.Pharm.Review, Vol. 10(3). 2007,104 - 109
2. J. Breitenbach, Drug delivery, a tool for imorived products: Kaletra® tablets, Presentation at Ìnforma Life Sciences Conference `Strategies to Enhance Solubility and Drug Absorption`, December 2007, Brussels
3. D. Bikiaris et al. Investigation of the release mechanism of a sparingly watersoluble drug rom solid dispersions in hydrophilic carriers based on physical state of drug, particle size distribution and drug-polymer interactions, Eur.J.Pharm.Biopharm 66 (2007) 334-347
4. ATM Serajuddin, Solid dispersion of poorly water-soluble drug: early promises, subsequent problems, and recent breakthroughs, .Pharm. Sci (1999), 88: 1058-1066
5. A. Fahr, X.Liu, Drug delivery strategies for poorly soluble drugs, Expert Opin. Drug Deliv. (2007) 4(4): 403 - 416
6. C. Timpe, A. Diederich, O. Kalb, A novel solvent-based fluidized bed wet granulation process for solid dispersions, Am.Pharm.Review, Vol. 11(4). 2008, 75 – 81
7. J. Jinno, N. Kamada, M. Miyake, K. Yamada, T.Mukai, M. Odomi, H. Toguchi, G.G. Liversidge, K. Higaki, T. Kimura, Effect of particle size reduction on dissolution and oral absorption of a poorly water-soluble drug, cilostazol, in beagle dogs, J. Control. Release 111 (1-2) (2006) 56 – 64
8. R.H. Müller, K. Peters, Nanosuspensions for the formulation of poorly soluble drugs I: Preparation by a size-reduction technique, Int.J.Pharma. 160 (1998) 229 – 237
9. E. Karavas, E. Georgarakis, M.P.Sigalas, K. Avgoustakis, D.Bikiaris , Investigation of the release mechanism of a sparingly water-soluble drug from solid dispersions in hydrophilic carriers based on physical state of drug, particle size distribution and drug–polymer interactions, European Journal of Pharmaceutics and Biopharmaceutics 66 (2007) 334–347.
10. D.Hörter, J.B. Dressmann ,Influence of physicochemical properties on dissolution of drugs in the, gastrointestinal tract, Advanced Drug Delivery Reviews 25 (1997) 3-14.
11. M. Wunderlich, R. Becker, Th. Bock, R.Brickl, W., J.Dressman, Predicting drug formulation effects on the precipitation of a poorly soluble basic drug in the small intestine, J. Pharm. Pharmacol.,2004,vol.56,no1,.43-51
12. K. Miyazaki, The use of an in vitro dissolution and absorption system to evaluate oral absorption of two weak bases in pH-independent controlled release formulations, Eur.J.Pharm. Sciences 26 (2005) 1–84.
13. G.G. Liversidge, K.C. Cundy, J. Bishop, D. Czekai, 1991.Surface modified drug nanoparticles, US Patent No. 5145684.
14. G.C. Liversidge, P. Conzentino, Drug particle size reduction for decreasing gastric irritancy and enhancing absorption of naproxen in rats, Int. J. Pharm. 125 (1995) 309–313.
15. G.C. Liversidge, K.C. Cundy, Particle size reduction for absolute oral bioavailability of nanocrystalline danazole in beagle dogs, Int. J. Pharm. 127 (1995) 91–97.
16. R.H. Mueller, C. Jacobs, O. Kayser, Advanced Drug Delivery Reviews 47 (2001) 3–19
17. Z. Deng et al., Understanding a relaxation behavior in a nanoparticlesuspension for drug delivery applications, International Journal of Pharmaceutics 351 (2008) 236–243
18. L. Lindfors et al., Amorphous Drug Nanosuspensions. 1. Inhibition of Ostwald Ripening, Langmuir 2006, 22, 906-910
19. J. Lee , J.-Y. Choi, C.H. Park, Characteristics of polymers enabling nanocomminution of water-insoluble drugs, Int. J. Pharm. 355 (2008) 328–336
20. B. Van Eerdenbrugh, G. Van den Mooter, P. Augustijns, Top-down production of drug nanocrystals: Nanosuspension stabilization, miniaturization and transformation into solid products, Int. J. Pharm. 364 (2008) 64–75
21. R.H. Müller, J. Möschwitzer, F.N. Bushrab, Manufacturing of nanoparticles by milling and homogenization techniques. In: R.B. Gupta, U.B. Kompella (ed.), Nanoparticle Technology for Drug Delivery, Drugs and the Pharmaceutical Sciences, vol. 159, Taylor & Francis Group, LLC (New York), 21 - 51
22. 22B. Van Eerdenbrugh, L. Froyen, J. Van Humbeeck, J. A. Martens, P. Augustijnsa, G. Van Den Mooter, Alternative matrix formers for nanosuspension solidification: Dissolution performance and X-ray microanalysis as an evaluation tool for powder dispersion, Eur.J. Pharm. Sciences 35 (2008) 344–353
23. Dong Z, Chatterji A, Sandhu H, Choi DS, Chokshi H, Shah N. 2008. Evaluation of solid state properties of solid dispersions prepared by hot-melt extrusion and solvent coprecipitation.Int J Pharm 335:141-149
24. T. Vasconcelos, Solid dispersions as strategy to improve solubility/dissolution of poorly soluble drugs (Presentation), 2nd Enhanced Solubility and Bioavailability IQPC Conference, Frankfurt 2009
25. J. Breitenbach et al., Confocal Raman Spectroscopy: Analytical Approach to Solid Dispersions and Mapping of Drugs, Pharm. Res. 16: 1109- 1113 (1999)
26. L. Yu, Amorphous pharmaceutical solids: preparation, characterization and stabilization, Adv. Drug Deliv. Rev., Vol. 48 (1), 2001, 27 – 42
Carsten Timpe, Ph.D., born in Hannover/Germany, studied Pharmacy at the Technische Universität Braunschweig. He completed his Ph.D. thesis at the Philipps University of Marburg/Germany in the group of Prof. Dr. Klaus Hartke in Pharmaceutical Chemistry (“Synthesis and Properties of novel 1.3-Dithiols with anellated Cyclopentadiene Ring”). From 1992 until 2000 he served as a group leader for formulation development of oral solid dosage forms (with ultra-low doses) at the Jenapharm GmbH in Weimar/Germany (affiliate of Schering AG). Subsequently he joined Eli Lilly in Hamburg/Germany in May 2000 where he worked as “Principal Research Scientist” until February 2007 in the formulation development area of new drug candidates with a special focus on technologies to overcome poor aqueous solubility of small molecules. He joined Novartis in Basel in 2007 as “Fellow” in the Special Delivery Systems group in Pharmaceutical Development and is currently working also as part-time project leader. Dr. Timpe is inventor/co-inventor of several international formulation patents and invited speaker to several drug delivery conferences in Europe and an active member of the APV focus group “Drug Delivery” in Germany.