Introduction
Utilization of prefilled syringes as a preferred container closure system for biologics has been increasing [1]. As a primary container closure system, prefilled syringes must provide an integral barrier that protects drug product stability and sterility throughout its entire shelf life. Drug manufacturers are required to check and demonstrate the system is capable of maintaining its microbial barrier integrity [2, 3]. In 2008, FDA further promoted container and closure system integrity (CCI) testing as a component of the stability protocol for sterile products
In response to the increasing regulatory expectations, the pharmaceutical industry has driven and witnessed significant technical advancements in CCI testing [5]. Instrumentation-based technologies, such as high voltage leak detection (HVLD) [6], vacuum/ pressure decay [7], mass extraction [8], and tracer gas detection (helium, oxygen etc.) [9, 10], have emerged and demonstrated improved detection capabilities compared to conventional dye and microbial ingress methods. Many of the technologies have been used for on-line 100% inspection and/or drug product stability CCI testing. In this article, we highlight our current thinking in an attempt to devise a systematic approach for CCI testing method selection, development, and validation.
General Considerations
In order to function both as a container closure system and as a drug delivery device, prefilled syringes feature many unique design elements. They usually include multiple containment compartments that are sealed by numerous interfaces. For example, the current stake needle glass syringes (Figure 1) provide a syringe barrel compartment for drug product containment and a separate needle shield compartment for needle protection. The syringe barrel compartment is sealed by the plunger on one end and by the needle on the other with the needle tip embedded in the needle shield. The needle shield compartment, sealed by the syringe barrel head,protects the needle exterior surfaces from potential contamination. The potential failure modes associated with each compartment and seal interface need to be identifi ed, assessed, and taken into account during CCI testing method development.
"In order to function both as a container closure system and as a drug delivery device, prefilled syringes feature many unique design elements.“
Furthermore, the plunger in a prefilled syringe is allowed to move within a range along the syringe barrel. When experiencing lower pressure environment during shipping and distribution, plunger movements in response to pressure variations may potentially aff ect seal integrity. Therefore, it is essential to evaluate plunger seal integrity following these special conditions.
In addition to the complex designs of prefilled syringes, the drug products packaged therein should also be considered. For example, prefilled syringes have been widely used for biologics, some of which could require extremely low temperature storage (e.g. -70°C). Since seal property of syringe components, especially elastomers (e.g. needle shields and plungers), is temperature dependent, CCI testing under extremely low temperatures could be required if theoretical justifications based on elastomer property are not adequate [11]. Moreover, drug-package interactions may impact method sensitivity and selection. For example, proteinaceous products could prevent mass transfer through CCI defects and reduce the sensitivity of a vacuum decay method [12].

Figure 1. Illustration of a stake needle glass syringe
CCI testing strategy for development
Many distinct CCI failure modes can occur throughout the life cycle of a syringe, ranging from component manufacturing, drug product filling and sealing, device assembling and packaging, to subsequent distribution and storage. It is essential to develop an overarching strategy to apply a series of CCI testing throughout the entire syringe life cycle.
CCI testing strategy development started with thorough understanding of syringe construction, design, and manufacturing processes. The CCI failure modes and eff ects associated with each aspect were first identifi ed. Using a risk-based approach, we further determined whether CCI testing is required, and if so, the intended uses and testing frequencies needed. For example, knowing the needle shield compartment seal integrity was tested by the component supplier, we elected to apply a non-routine CCI test to confirm its seal integrity upon drug product filling and sealing, and upon being assembled into devices. In contrast, for the product-containing syringe barrel compartment, we incorporated an extensive set of CCI tests into the entire product development cycle, including initial design confirmation, machinability studies, and product stability testing, to ensure CCI was achieved and well maintained.
Method selection
Table 1 lists the major CCI testing technologies available for prefilled syringes and their key characteristics. Note all the technologies have major limitations. When selecting appropriate methods, the following key aspects should be considered.
- Suitable for its intended use. The selected method(s) must be suitable for the intended use and scope of a specific CCI test. For example, microbial ingress testing, although a good selection for media-filled syringes for fil lling process validation, cannot be used for stability testing because it does not apply to drug product filled samples. If a single method cannot meet all the testing needs, complementary methods may be applied in tandem to achieve definitive and comprehensive testing conclusions.
- Applicable to the specific drug product-package. As previously mentioned, drug products can interact with CCI defects in various ways and may further aff ect the eff ectiveness of CCI testing methods. The method applicability to the specic product-package must be evaluated and adequately demonstrated.
- Detection capability and eff ectiveness. Recent technologies utilizing mass extraction [8], HVLD [5], vacuum decay [7], have demonstrated reliable detection of CCI defects of 5-10 microns or smaller. These technologies are based on quantitative measurement of certain sample characteristics that can be further correlated to presence and/or sizes of CCI defects. The superior sensitivity and reliability made them preferred CCI testing methods over conventional dye or microbial ingress tests.
- Non-destructive CCI testing. Non-destructive methods enable 100% CCI testing. In addition, they allow for further analysis of the failure modes and root causes, which in-turn provides valuable feedback for continuous improvement.
Method development
Upon establishing a preliminary method following vendor’s recommendations or literature search, we further focused on optimizing testing parameters and determining the appropriate pass/fail threshold.
Optimize Testing Parameters
First, various defect standards of known sizes (Table 2) were tested along with intact samples under different testing parameters. The correlations were thoroughly explored between key method parameters and instrument responses to intact and defect samples, aiming to identify a set of parameters that yield optimized separation between defect and intact samples (i.e. signal-to-noise ratio).
Refine Pass/Fail Threshold
To establish the preliminary pass/fail threshold, the optimized method was used to test multiple lots of filled intact syringes representing relevant product variations, including various packaging component sources/lots, drug products batches, as well as packaging sites and lines. The testing results were statistically evaluated to define the instrument baseline and variation (σ) for intact samples. Ideally, the pass/fail threshold should be at 10σ above baseline (i.e. above limit of quantitation LOQ). Defect standards of known sizes were then tested to further finalize and verify the pass/fail threshold. In cases where the 10σ threshold did not provide the desired sensitivity (as illustrated in Figure 2), the threshold setting was further adjusted between 3σ above baseline (i.e. limit of detection LOD) and the 10σ LOQ to achieve the desired detection sensitivity while keeping false positive detection probability (i.e. intact sampled detected as Fails) within the acceptable level.
Verify Method Effectiveness
Although defect standards are essential for initial method definition and optimization, they do not necessarily fully represent natural CCI defects. Natural CCI defects are of a large variety and most of them are not simple orifices or tubes. Therefore, the method performance was further evaluated using “real-world” CCI defects.
A good “real-world” defect sample set should represent all major probable CCI failure modes. Actual CCI defects could be obtained from various sources, such as reject samples from incoming or inprocess controls. When actual defect samples were not available for a specific failure mode and defect type, simulated defects were used.
A few iterations of the steps above may be needed to finalize the method. For methods used for stability testing, additional studies were performed to verify the methods are capable of detecting “aged” samples. Usually it was demonstrated by placing a set of productfilled samples with known defects on a stability study and testing the defect samples at various time points.
Method validation
Table 1. Characteristics of Major CCI Testing Methods

Table 2. Commonly-used CCI Defect Standards
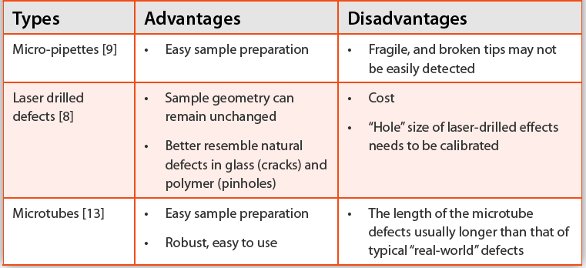
In general, ICH analytical method validation guideline [14] was followed to validate instrument-based CCI testing methods. The key method characteristics, such as detection limit, range, accuracy, precision and robustness, were evaluated and demonstrated during the validation stage. In order to demonstrate detection capability in size, micro-pipettes, microtubes, and laser drilled standards of known sizes were usually used, which also allowed direct comparison of testing capability of various methods.
CCI testing methods were validated for the specific drug productpackage. Because the drug product formulation and package design may change during early development phases, a phase-appropriate approach was implemented to validate methods in concert with product development phases. For example, we utilized scientifically sound methods to support packaging system qualification and development stability studies. Once the product formulation and packaging design were finalized, the methods were then fully validated in support of primary stability and process validation CCI testing. Additional long-term method robustness may be further validated prior to implementing the method in QC laboratories for routine testing.
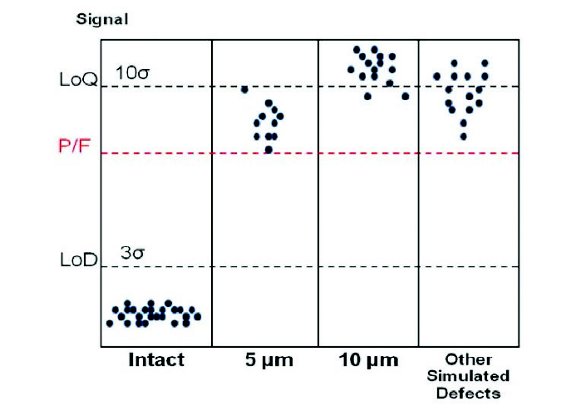
Figure 2. Approach to establishing Pass-Fail threshold
Summary
Appropriately selected and validated methods are essential for demonstrating container closure integrity during package and drug product development and manufacturing. However, it should be realized that current CCI testing technologies do not off er an ideal method that satisfy all prefilled syringe CCI testing needs. An integrated approach incorporating CCI testing and other engineering and administrative controls must be taken to ensure overall container closure integrity.
Acknowledgements
The author wishes to thank Jon Parker for providing the syringe drawing and Ross Allen, Yu Hu, and Randy Thackrey for helpful comments and review of this article. Discussions with Seungyil Yoon, Craig Goldhammer, Dana Guazzo, and Heinz Wolf on CCI testing technologies are greatly appreciated.
Author biography
Lei Li, Ph.D., is currently Associate Senior Consultant Engineer at Eli Lilly and Company. He has been with Eli Lilly since 2009 in packaging development with a focus on container closure system design, characterization, and qualification. He obtained a Ph.D. in Analytical Chemistry from West Virginia University and worked as an analytical scientist at GE Plastics prior to joining Lilly.
References
- Kiang P. Recent Developments in Prefilled Syringes, American Pharmaceutical Review, September/October 2012: 104-107
- US State Food and Drug Administration (1993). FDA 1993 Guidance for Industry for the Submission Documentation for Sterilization Process Validation in Applications for Human and Veterinary Drug Products
- EU Guideline to Good Manufacturing Practice (2008). Medicinal Products for Human and Veterinary Use, Annex 1. Manufacturer of Sterile Medicinal Products.
- US State Food and Drug Administration (2008). Container and Closure System Integrity Testing in Lieu of Sterility Testing as a Component of the Stability Protocol for Sterile Products
- Li L. Container Closure Integrity Testing Method Development and Validation for Pre-filled Syringes, PDA 2012 Universe of Pre-filled Syringes & Injection Devices Conference, October 16, Las Vegas, Nevada
- Möll F. et al. Validation of a High Voltage Leak Detector for Use with Pharmaceutical Blow-Fill-Seal Containers – A Practical Approach, PDA J Pharm Sci and Tech, 1998, 52, 5 215-227
- Wolf H. et al. Vacuum Decay Container/Closure Integrity Testing Technology. Part 2. Comparison to Dye Ingress Tests, PDA J Pharm Sci and Tech 2009, 63, 489-498
- Yoon S. et al. Mass Extraction Container Closure Integrity Physical Testing Method Development for Parenteral Container Closure Systems, PDA J Pharm Sci and Tech, 2012,66 403-419
- Kirsch L. et. al. Pharmaceutical Container/Closure Integrity I: Mass Spectrometry-Based Helium Leak Rate Detection for Rubber-Stoppered Glass Vials, PDA J Pharm Sci and Tech, 51, 5 187-194
- Hirotaka S. et al. Development of a Nondestructive Leak Testing Method Utilizing the Head Space Analyzer for Ampoule Products Containing Ethanol-Based Solutions, PDA J Pharm Sci and Tech, 2012,66 434-444
- Brigitte Z. et al. Container/Closure Integrity Testing and the Identification of a Suitable Vial/Stopper Combination for Low-Temperature Storage at −80 °C, PDA J Pharm Sci and Tech, 2012,66 453-465
- Orosz, Jr. S.; Guazzo D. Glass Vial Finish Defects: Leak Detection and Product Risk Assessment. PDA Annual Meeting, Packaging Science Interest Group, March 16, 2010 Orlando, FL
- Keller S. Determination of the Leak Size Critical to Package Sterility Maintenance, Dissertation submitted to the faculty of Virginia Polytechnic Institute and State University, 1998
- ICH Guidance for Industry, Q2B Validation of Analytical Procedures: Methodology, 1996