Introduction
The dissolution method is a critical part of oral drug product development. It provides the most intimate link between material properties, formulation design, process influence and the ultimate performance in the patient. The information that the team needs from the dissolution method changes as a project advances in time. At all stages of development, a properly designed dissolution method allows the drug product development team to understand and assess the risks that exist in their drug product, to develop a robust formulation and process that minimizes these risks and to identify important changes in the drug product during its manufacture.
In order to develop a dissolution method with a sound basis, some fundamental characterization work must be completed. Since the basic process of dissolution involves converting the solid particles of interest from their crystalline or amorphous matrix into individual ions, atoms or molecules and then transporting them into the solvent, the solid form of the solute compound is of great interest. Early in development (pre-phase II) work is undertaken to understand the acceptable API attributes for the formulation and API which have been selected. This work is crucial to enable the team to develop useful dissolution methods that are able to inform the formulation decisions that will be made later. The fundamental understanding of the solid forms of the compound should not only include definition of the reference solid form, but also of those related forms (especially those that are less soluble) which may have some influence on dissolution. Additionally, work should be undertaken to understand the impact of other API attributes such as particle size, crystallinity and any other properties deemed to be important, as these can have profound impact on dissolution performance as well. In this early phase of development, the dissolution method is used to explore and understand the impact of various parameters on the performance of the formulation. As development proceeds and more is learned about the drug product,a connection to the performance that will be observed in-vivo is desired. Finally, in later stages of development, the dissolution method transforms to become one which is more rigid and which is used to evaluate the consistency of the manufacturing process, material properties and drug product performance.
Late Stage Dissolution Methods
There are many options to consider when developing a dissolution test. The nature of the solvent (the undersaturation), the type of apparatus used and the amount and type of mixing are various factors that impact the ability of the dissolution method to discriminate between dosage forms. The type of formulation and its intended mechanism may also influence the design of the dissolution method. Although there may be several choices of conditions which will allow the basic quality of the formulation to be assessed, only through careful understanding of the dissolution mechanism and the materials involved can a dissolution method be developed that will identify the likely problems that can occur with the formulation.
The importance of the conditions selected for a late stage dissolution method can be illustrated by considering two examples. Delavirdine mesylate [1] and prasugrel hydrochloride [2] are both formulated as immediate release tablets and both have similar solubility properties. They are water soluble salts of very insoluble basic compounds formulated as tablets. Despite the similarities of the compounds, the dissolution methods developed for each tablet were very different. The dissolution method developed for delavirdine mesylate tablets used a high pH condition (with surfactant added) whereas the dissolution method for prasugrel hydrochloride tablets used a low pH media. This choice was important to identify problems with the formulation late in the development. During the development of delavirdine mesylate, a slowdown in the dissolution profiles during accelerated stability studies was observed as shown in Figure 1.
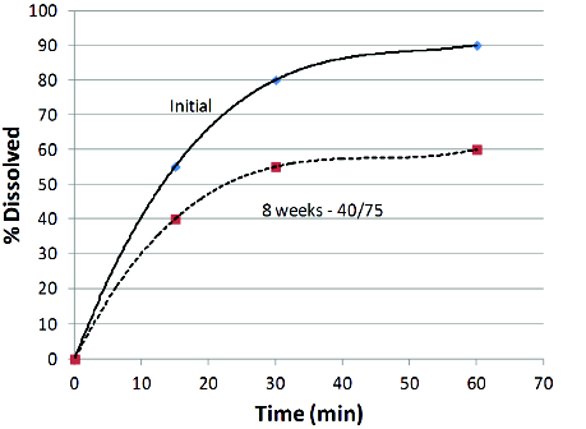 Figure 1. Reduction in the dissolution rate for delavirdine mesylate tablets observed after 8 weeks on accelerated stability at 40C/75%RH (open).
Figure 1. Reduction in the dissolution rate for delavirdine mesylate tablets observed after 8 weeks on accelerated stability at 40C/75%RH (open).After some work, the authors were able to correlate this change in dissolution with a solid-state chemical reaction between absorbed water, the croscarmellose sodium disintegrant and the delavirdine mesylate to produce highly insoluble delavirdine free base, which slowed the dissolution. The identification of this issue allowed improvements to the formulation to be made. In the case of prasugrel hydrochloride, a very similar conversion to the free base form had occurred, but because the dissolution method was developed at low pH, it was non-discriminating for the solid form of the active, therefore, the conversion to the free base (between 42-87%) failed to be discovered until pivotal clinical trials were already underway. As one might expect this led to questions about bioequivalence, especially in patients that might be using antacids, late changes in drug product and, in this case, post marketing commitments to alter the formulation (as well as the dissolution method) to satisfy regulators [3]. Obviously, early discovery of this problem would have eased the formulation development process significantly.
Early development-Inherent API Properties
As the previous examples showed, the early stage work in developing an understanding of materials and their behavior as well as the understanding the release mechanism from the formulation is important to the development of an effective late stage dissolution method. Early dissolution methods may focus on understanding the API properties, or on more fundamental dosage form properties, such as disintegration or maintenance of supersaturation. The solid form landscape needs to be well understood in order to design an effective dissolution test. The solid phases that might result from changes in the dosage form either prior to dosing or during dosing should be considered. Supersaturation levels are typically important to measure in order to understand how they impact dissolution performance. Early in development, intrinsic dissolution testing is a good method to build an understanding of the fundamental limits of the API itself.
In its simplest manifestation, intrinsic dissolution testing can be used to assess the BCS class of a compound [4]. However, more detailed studies of precipitation and solid form conversions can also be completed [5]. These can help to define the best ways to formulate the compound and to determine the risk factors for changes in dissolution performance later in development. A simple example is shown in Figure 2. Here, rotating disk dissolution profiles for a hydrochloride salt of a weak base compound as a function of pH are shown. In this case, the dissolution proceeds in a linear manner, as would be expected, at pH 2. However, at higher pH values, the dissolution drops off significantly. This is caused when precipitation of the free base form of the compound occurs. The free base solid form has solubility much less than that of the salt, leading to depressed dissolution. Knowledge of the propensity of this occurrence and the pH at which it occurs is critical to designing an effective formulation for this compound.
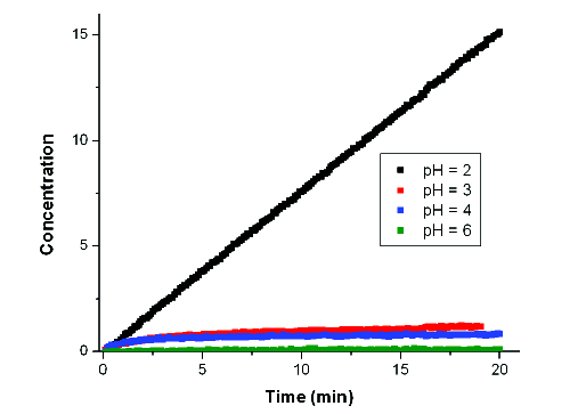 Figure 2. Rotating disk dissolution data for a hydrochloride salt of a poorly soluble base demonstrating the impact of pH on the dissolution performance.
Figure 2. Rotating disk dissolution data for a hydrochloride salt of a poorly soluble base demonstrating the impact of pH on the dissolution performance.Another example of understanding dissolution fundamentals can be illustrated by examining co-crystals. Co-crystals, in some cases, have been able to show enhanced dissolution, but one concern is always dissociation of the co-crystal into its individual components, destroying the advantage. In one published example [6], the theophylline/acetaminophen co-crystal was studied and the rotating disk intrinsic dissolution data is shown in Figure 3. This data shows that the formulation of a co-crystal does indeed enhance the dissolution rate significantly. Furthermore, by independently measuring the appearance of both the theophylline and acetaminophen, the authors were able to assess that the cocrystal was dissolving congruently as a co-crystal and not simply dissociating into its parent compounds. Developing fundamental understanding of the dissolution of the API early in development will lead to the development of better, biorelevant dissolution methods later on.
 Figure 3. Rotating disk dissolution data for theophylline/ acetaminophen 1:1 mixtures and co-crystal showing enhanced dissolution rate of the co-crystal (measured by theophylline concentration).
Figure 3. Rotating disk dissolution data for theophylline/ acetaminophen 1:1 mixtures and co-crystal showing enhanced dissolution rate of the co-crystal (measured by theophylline concentration).Middle development - Biorelevant testing
As a compound progresses further into development there is typically a desire to predict in-vivo performance accurately. The concepts behind the BCS system demonstrate how scientific thought should govern the development of dissolution methods in order to predict bioperformance [7]. Standard USP II type dissolution testing can be used to assess certain compounds, but others require more advanced test methods. In the recently completed AAPS workshop on 21st Century Oral Bioperformance Prediction and Testing, there were several topics addressed. These included standard USP testing, but it was recognized that the USP II test is limited in its scope and the conference also covered many other topics such as biphasic dissolution, membrane permeation models, processes for integrating in-vitro and in-silico models, dissolution with more biorelevant media and using more complex dissolution systems. Once the fundamentals of a compound and its formulation are understood, these more biorelevant methods can be applied to both build understanding of the formulation performance and to predict in-vivo performance. For highly soluble and permeable compounds, most of these techniques do not need to be applied and a simple USP II method will be sufficient, but, for BCS II or IV type compounds biorelevant methods can be very powerful.
One example of the utility of these alternate methods will be presented here as an illustrative example of how these approaches can be applied to assist in the design of a formulation [8]. In this case, the compound of interest was a pain reliever and a simple formulated capsule was designed to be dosed in the clinic in a Phase II efficacy trial. The compound was a BCS II acid with a pKa of about 3.3 and a very poor intrinsic solubility of less than 1 microgram/mL. Despite the poor solubility at low pH, the capsule formulation’s dissolution properties were deemed acceptable under sink conditions at higher pH. Because of this, the prediction was that the AUC of the capsule formulation would be equivalent to that of the solution formulation which had been dosed in earlier clinical studies and efficacy would be acceptable. Unfortunately, the clinical study showed that the compound failed the efficacy testing because the time required to reach the critical concentration needed for pain relief was highly variable [9]. Therefore, a reformulated dosage form was required which would provide solution-like performance from a solid oral dosage form. The main challenge to achieving this goal was that the compound was poorly soluble in the low pH stomach in which it needed to dissolve rapidly. This fact also made it difficult to develop a dissolution test since the standard, sink condition type dissolution tests were not representative of the in-vivo environmental conditions.
To solve this problem, several dissolution methods were tested, but the artificial stomach-duodenum method (ASD) appeared to provide the best results [10]. This method incorporates pH changes in a dynamic manner and is more representative of the limiting factors that are present in this case. The team made several diff erent formulations of the compound and tested them in the ASD system. The key criteria was the concentration in solution at t=30 min as determined by the PK modeling available for the compound [9]. ASD runs were completed for several formulation approaches and three solid formulations were selected based on the ASD results (Dudodenal AUC from 0 to 30min) to test in a human PK study. These formulations were selected since they showed a range of behavior in the in vitro test. The results of the ASD and human PK study are shown in Figure 4. In this example, a nice correlation was observed between the ASD predictions and the actual human results for exposure. The PK modeling was then able to confirm that the best performing formulation would be sufficient to obtain the desired result in the clinic. This was subsequently confirmed with a second efficacy trial with the new formulation. In this example, the biorelevant test method was critical to assist in development of the formulation and building an understanding of which mechanisms were most eff ective for this compound. These types of formulations would have been much more difficult to screen using more traditional dissolution approaches.
 Figure 4. Correlation between ASD AUC and Human PK data for three solid, oral formulations as compared to solution formulation.
Figure 4. Correlation between ASD AUC and Human PK data for three solid, oral formulations as compared to solution formulation.Conclusion
Dissolution testing is a critical tool to enable the design of effective drug products. A properly designed late stage dissolution method is useful in identifying problems with the formulation during its lifetime. Earlier stage dissolution tests are built around understanding the physical performance of the API and the formulation. The knowledge gained from these early tests will define how to design the late stage test method in order to successfully monitor the performance of the product.
Acknowledgements
Thanks go to numerous collaborators over the years whose collective thoughts are inherently included in this paper. Additional thanks to Mike Meyers and Anjali Agrawal for helpful comments on this paper.
Author Biography
Michael Hawley, Ph.D., received his BS in Chemistry from Binghamton University and Ph.D. in Physical Chemistry from the University of Arizona in 1991. His experience in the pharmaceutical industry began with The Upjohn Company in 1994. Michael is currently leading the Solid Formulation Development group within the Pharmaceutical Development organization at Boehringer-Ingelheim at the Ridgefield, CT site and has co-authored over 40 publications and patents with various collaborators.
References:
- B.R.Rohrs, T.J.Thamann, P.Gao, D.J.Stelzer, M.S.Bergren, R.S.Chao. Tablet dissolution affected by a moisture mediated solid-state interaction between drug and disintegrant. Pharm Res. 16(12), 1850-6 (1999).
- E.F.Unger. Prasugrel for Reduction of Cardiovascular Events in Patients with Acute Coronary Syndrome (ACS). Cardiovascular and Renal Drugs Advisory Committee. Silver Spring, Maryland. 3 February 2009.
- M.J.Laffler, C.Werble. Effient approval was prolonged, but FDA requirements may help sponsors. Pink Sheet. Elsevier Business Intelligence. 21 July 2009.
- L.X. Yu, A.S. Carlin, G.L. Amidon, A.S. Hussain. Feasibility studies of utilizing disk intrinsic dissolution rate to classify drugs. Int. J. Pharm. 270 (1–2), 221–7 (2004).
- M.Hawley, W.Morozowich. Modifying the Diffusion Layer of Soluble Salts of Poorly Soluble Basic Drugs to Improve Dissolution Performance. Mol. Pharm. 7(5), 1441–9 (2011).
- H.G. Lee, G.G.Z.Zhang, D.R.Flanagan. Cocrystal Intrinsic Dissolution Behavior Using a Rotating Disk. J.PharmSci., 100, 1736-44 (2011).
- G.L.Amidon, H.Lennernas, V.P.Shah, J.R.Crison. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 12(3), 413-20 (1995).
- M.Hawley. Application of Rotating Disk Dissolution to Solid State Science. Dr. David W. Grant Symposium - Solid State Pharmaceutics: Present and Future. Minneapolis, MN (2010).
- K.G.Kowalski, S. Olson, A.E.Remmers, M.M.Hutmacher. Modeling and Simulation to Support Dose Selection and Clinical development of SC-75416, a selective COX-2 Inhibitor for the Treatment of Acute and Chronic Pain. Clin.Pharm.Ther. 83(6), 857-66 (2007).
- M.McAllister. Dynamic Dissolution: A Step Closer to Predictive Dissolution Testing? Mol. Pharm. 7(5), 1374-87 (2011).