Introduction
In the past decade, packed column supercritical fluid chromatography (SFC) has dominated chiral separation and chiral purification in medicinal chemistry and process chemistry in the pharmaceutical industry. However, SFC is not just limited to chiral separations. It is getting more and more attention in the arena of achiral separation, which has been dominated by reversed-phase liquid chromatography (RPLC). Compared to well-established RPLC, SFC provides higher separation efficiency and faster analyses while offering low operation cost and facile tunable selectivity for method development. More attractively, SFC offers complementary selectivity to RPLC, making itself a valuable tool in the analyses of complex mixtures comprised of analytes which may be structurally related but range in polarity. In this review article, the authors describe their practical approaches for developing achiral analytical SFC methods and discuss their strategies of implementing SFC in drug development for the testing of drug substances and formulated dosage forms.
Achiral SFC Method Development
Stationary Phase Screening
Due to lack of knowledge of the interactions between the analytes and the SFC system, there are few clear guidelines on column selections for SFC method development [1]. In practice, multiple columns need to be examined in order to identify a suitable stationary phase. One way to simplify the column selection is to classify the stationary phases based on their potential retention mechanism and then choose one or two representative columns from each category for screening. Once a hit is identified, more columns in that particular class can be further scrutinized.
West and Lesellier have constructed comprehensive evaluations for commercially available SFC columns using QSRR (quantitative structure-retention relationship) approach [1-6]. As shown in Figure 1, they developed a five-dimensional classification diagram based on a linear solvation energy relationship, grouping the columns by retention similarity. Three major groups of stationary phases can be defi ned: nonpolar, moderately polar and very polar [1]. The first group is constituted by alkyl-bonded stationary phases that possess no polar groups. With the non-polar phase, SFC behaves more like reversed-phase retention mode. The second group is constituted by stationary phases of intermediate polarity, and comprises all ODS phases containing a polar-embedded group or a hydrophilic endcapping group, along with a wide variety of aromatic stationary phases. With the stationary phase in this group, SFC behaves in a mixed mode that is in between reversed-phase retention mode and normal phase retention mode. The third group is constituted by polar stationary phases. This group comprises a fluoroalkyl phase (FD), polar polymer-coated phases (PEG and PVA), the ethylpyridine bonded silica (EP) along with more classical phases such as bare silica gel (Si), aminopropyl-bonded silica (NH2), cyanopropyl-bonded silica (CN) and propanediol-bonded silica (Diol). In this, SFC behaves like normal phase retention mode with the polar phase. It is recommended that the initial column screening is started with picking one or two columns from each group. These stationary phase combinations would provide a high probability of finding the appropriate starting conditions for method optimization.
 Figure 1. Spider diagram for a fi ve-dimensional representation of the SFC column selectivities [6]
Figure 1. Spider diagram for a fi ve-dimensional representation of the SFC column selectivities [6]Role of SF CO2 Modifier and Additive
Supercritical fluid CO2 is no more polar than hexane and its elution strength is too weak to elute most polar compounds. Its solvent strength can be increased by adding small volumes of polar organic solvents. Use of these modifiers can influence the SFC separation by changing analyte interaction, mobile phase, or stationary phase, as described:
For the analyte, the modifier may selectively solvate polar compounds to form clusters with different distribution properties [7].
For the mobile phase, the modifier can alter the density and solvating power of the mobile phase.
For the stationary phase, adding a modifier has multiple impacts [8]:
- Modifier can block active sites on the stationary phase;
- Adsorbed modifier can increase the volume of the stationary phase leading to a change in the column phase ratio;
- Adsorbed modifier can act as a component of the stationary phase.
Even with a polar organic solvent as a modifier, SFC is still not always sufficient to facilitate elution in a reasonable time or with an acceptable peak shape for some ionic compounds and polar compounds. A third more polar component (e.g., additive) added into the mobile phase can help to mitigate this problem. Suggested roles for additives in the separation process include [9-13]:
- enhancing mobile phase solvating power;
- suppressing ionization of ionic analytes;
- ion-pairing with ionic analytes;
- modifying the stationary phase.
Typical additives are strong acids, bases or salts. In some instances, a small percentage of water is also used as an additive to help elute highly polar compounds, such as nucleobases and polypeptides, where water introduces HILIC-like analyte partitioning [14].
Fine-tuning of SFC Separation
Like HPLC, the SFC mobile phase strength is the function of the mobile phase composition, by the nature of modifiers and their concentrations. What is unique to SFC is that the mobile phase strength is also a function of the density. This feature gives SFC additional means to adjust retention and selectivity. In fact, SFC method development can be divided into two parts: “primary” screening by searching the stationary phase and mobile phase, and “fine” tuning by optimizing the separation temperature, pressure, and flow rate. The fine tuning is best performed after selection of the best stationary phase and mobile phase composition for a given application.
Changing the separation pressure alters the density of the fluid and as a result, changes the relative retention of the analytes. Increasing pressure will increase the fluid density and the solvation strength, therefore resulting in shorter retention for the analytes. However, the magnitude of the changes in retention due to variation in pressure is small compared with the changes due to modifier concentration. However, pressure changes tend to produce larger changes in selectivity than does modifier concentration adjustment, although the diff erences are subtle [15]. Brunelli et. al. pointed out that the change in mobile phase density with the pressure had some infl uence on compound retention for less polar compounds, while such a diff erence in density did not alter the polarity of the mobile phase signifi cantly enough to infl uence more polar compounds [16]. In other words,for the SFC separation with a high percentage modifier, the effect of pressure on retention is greatly reduced. It also means that the pressure variation normally has more influence on the early eluting compounds than on those late eluting compounds.
Changing the temperature of the SFC mobile phase, at constant pressure, also changes the density of the fluid. On one hand, increased temperature causes a decrease in fluid density, and therefore increases the retention. On the other hand, increased temperature can also cause desorption of both CO2 and any modifiers from the stationary phase, which tend to reduce the retention [15]. Changing temperature also alters the kinetic energy of the solute, therefore, the impact of separation temperature on retention is hard to predict. This should be evaluated case by case.
One thing should be mentioned, flow rate is not a typical parameter for selectivity tuning as it has no direct influence on selectivity. Due to the low viscosity of supercritical fluid and a “flat” HETP-flow rate curve, flow rate is a useful tool in SFC to reduce the analysis time without compromising the efficiency. However, the system pressure increases when flow rate is increased. As a result, fluid density and mobile phase solvation strength alter, which consequently causes subtle shift in peak relative retention.
For samples with a complex matrix, e.g., formulated dosage forms, the sample preparation is a critical step in the overall analytical method development because this directly influences whether accurate and representative data are being generated. A detailed discussion on appropriate SFC sample prep strategies will be given in a later section of this article.
Developing SFC Method for Mometasone Furoate and its Related Compounds
To demonstrate the principles elucidated above, a step-by-step SFC method development is presented for the separation of mometasone furoate related compounds (e.g., process impurities and degradation products) [17].
A primary screening is conducted first to identify the best stationary and mobile phase. Since mometasone furoate and its impurities are relatively low polar compounds, three columns from the polar stationary phase group were selected: including 2-ethylpyridine, cyano, and silica. For the initial screening, neat methanol was used as the modifi er. Considering the analytes are neutral and non-ionic compounds, no additive was used. A shallow gradient (5% to 15% methanol) was used to screen these columns to separate the spiked mixture. The best separation was achieved on the silica column (Figure 2c) with all nine components baseline separated within 12 minutes. Further method development was then focused on the silica column and several modifiers were evaluated. For this study, methanol, ethanol, isopropanol, and acetonitrile were compared as potential modifiers and methanol was identifi ed to be the best modifier.
 Figure 2. Column screening on (a) 2-ethylpyridine, (b) cyano, and (c) silica columns. CO2 100 bar, 4.0 mL/min, 30°C, modifi er: methanol 5% to 15% in 15 min.
Figure 2. Column screening on (a) 2-ethylpyridine, (b) cyano, and (c) silica columns. CO2 100 bar, 4.0 mL/min, 30°C, modifi er: methanol 5% to 15% in 15 min.After the stationary phase and mobile phase were identifi ed, the second step of the method development was to fine-tune the separation by adjusting pressure and temperature. System back pressure at 100 bar and 150 bar were evaluated (Fig. 3a and 3b, respectively). The resolution was compromised at higher system pressure and compound 1 was only partially separated from compound 6. Temperatures of 30°C, 35°C, and 40°C were evaluated where the pressure was maintained at 100 bar (Fig. 3a, 3c, and 3d, respectively). In this case a better resolution is obtained at a lower temperature.
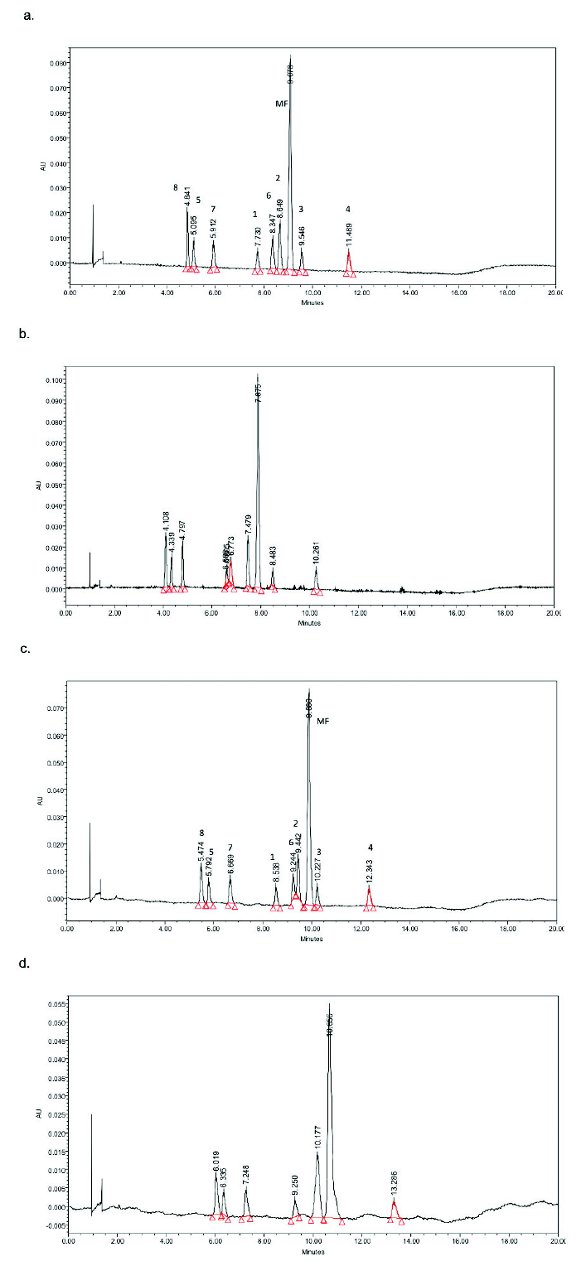 Figure 3. Evaluate mobile phase pressure and column temperature impact on selectivity. (a) 100 bar, 30°C; (b) 150 bar, 30°C; (c) 100 bar, 35°C; (d) 100 bar, 40°C. 4.0 mL/min, modifi er: methanol 5% to 15% in 15 min, silica column
Figure 3. Evaluate mobile phase pressure and column temperature impact on selectivity. (a) 100 bar, 30°C; (b) 150 bar, 30°C; (c) 100 bar, 35°C; (d) 100 bar, 40°C. 4.0 mL/min, modifi er: methanol 5% to 15% in 15 min, silica columnThe elution order of RP-HPLC and SFC was compared in Figure 4. Due to a polar stationary phase, e.g. silica, used for this application, the SFC demonstrated a normal phase- like retention mechanism. SFC has a dramatically different retention order compared to that of RP-HPLC, which is a very attractive feature for pharmaceutical impurity profiling.
 Figure 4. The orthogonal selectivity of SFC method vs. RP-HPLC method. SFC condition: 100 bar, 30°C, 4.0 mL/min, modifier: methanol 5% to 15% in 15 min, silica column. RP-HPLC condition: 25°C, 1.5 mL/min, water/acetonitrile (58:42, v/v) to water/ acetonitrile (48:52, v/v) in 60 min, Ultrasphere ODS column.
Figure 4. The orthogonal selectivity of SFC method vs. RP-HPLC method. SFC condition: 100 bar, 30°C, 4.0 mL/min, modifier: methanol 5% to 15% in 15 min, silica column. RP-HPLC condition: 25°C, 1.5 mL/min, water/acetonitrile (58:42, v/v) to water/ acetonitrile (48:52, v/v) in 60 min, Ultrasphere ODS column.SFC sample preparation strategy for Formulated dosage Forms

Despite its great success in medicinal chemistry and process chemistry labs, SFC has rarely been used for formulated dosage forms, e.g., drug product testing, where RPLC is the work horse. The main obstacle of implementing SFC in this arena is the potential challenge of sample preparation. Due to the unique physical properties of supercritical CO2, the preferred sample solvent for SFC analysis is neat organic. The samples in medicinal chemistry and process chemistry labs are typically starting materials, intermediates, or fi nal products of the chemical synthesis. Therefore, SFC is a natural fit for synthetic chemical analysis where the samples can be directly injected into SFC or after simple dilution with organic solvents. The sample matrixes of formulated drug products can be quite complicated, depending on the excipients and dosage forms. In drug product testing lab, as a critical part of analytical method development, sample preparation strategies are extensively studied to ensure that the sample treatment is representative and quantitative. The sample preparation is also required to be RPLC compatible, and as a result, an aqueous phase is used in the sample solvent. However, many sample preparation methods suitable for RPLC cannot be used for SFC because direct injection of aqueous is problematic [18] due to:
- Sample freeze during depressurization in the SFC back pressure regulator
- Sample precipitation out of solution when encountering the SFC polar organic mobile phase
- Extremely asymmetric and distorted peaks due to the column surface activity generated by the injected water in the column
The sample preparation that is suitable for SFC must be considered and evaluated. The proposed strategies for different dosage forms are summarized in Table 1.
Table 1. SFC sample preparation strategy for formulated drug product

For solid dosage forms (e.g. tablets and capsules) the sample preparation methods could be straight forward [19-24]. The powders (ground first for tablets) can be mixed/dissolved with organic solvent (such as methanol, ethanol, or acetonitrile) and then centrifuged or filtered before SFC injection.
For liquid dosage forms (e.g. ,aqueous solution, emulsion, or suspension), sample preparation could be much more challenging for SFC analysis than for RPLC analysis. Several researchers reported direct SFC injection of aqueous samples, which required replacing methanol with 2-propanol in the mobile phase and/or inclusion of an additive (e.g., 1mM citrate) in the organic modifier [25-28]. Mukherjee et.al. reported successful direct injection of aqueous formulation on chiral SFC [18,29]. The authors believe that the low injection volume and high nozzle temperature helped to mitigate the compatibility problem. For sure, direct injection of large volume of aqueous formulation is not recommended for SFC. Appropriate sample treatment, such as solid phase extraction (SPE) or liquid liquid extraction (LLE) can be considered. For instance, after failure of direct SFC injection of mometasone furoate aqueous formulation, SPE was utilized in our lab to convert aqueous matrix to organic. With the optimized combination of absorbent and elution solvent, quantitative recovery was achieved, as shown in Figure 5.
 Figure 5. SPE sample treatment prior to SFC injection (SPE-SFC): extraction recovery study of mometasone furoate aqueous formulation
Figure 5. SPE sample treatment prior to SFC injection (SPE-SFC): extraction recovery study of mometasone furoate aqueous formulationConcluding remarks
Reversed-phase HPLC has been established as the main work horse for impurity analysis during drug development and production. Having a normal phase-like retention mechanism, packed column SFC provides orthogonal selectivity that is complementary to RP-HPLC. In addition, SFC provides higher separation effi ciency and faster analyses with less consumption of organic solvents. Despite all these advantages and its popularity in chiral separation within pharmaceutical companies, SFC has not been fully utilized in achiral analysis for drug substance testing, and even rarely seen for drug product analysis. The major concern of implementing SFC to formulated dosage form testing is that the sample preparation needs to be carefully developed to be SFC friendly. For oral dosage forms, the sample preparation could be relatively straightforward for SFC analysis. For aqueous base dosage forms, some sample treatment may be required before SFC injection. With appropriate sample preparation put in place, SFC should gain wider acceptance in pharmaceutical analysis, not only for drug substance but also for drug product.
Author biographies
Dr. Zhenyu Wang currently works within Pharmaceutical Sciences and Clinical Supplies of Merck Research Laboratories in Summit, NJ. Previously, he was a Principal Scientist at Hoff mann-La Roche Inc. from 2005-2007, leading its analytical separation group in Discovery Chemistry Department. He received his Ph.D. in Analytical Chemistry at Virginia Tech, under the supervision of Prof. Larry Taylor. Zhenyu’s research interests include separation science, inhalation product analysis, sample preparation, and supercritical fl uid technology. He has over 20 peer-reviewed publications, two book chapters and one patent.
Honggen Zhang, M.S., received his B.S. in Chemical Engineering from Dong Hua University in Shanghai, China in 1977 and M.S. in Chemical Engineering from the University of Laval in Quebec, Canada in 1992. His experience in the pharmaceutical industry began with ICN (Canada) Company in Montreal, Canada in 1992. Honggen is currently a senior scientist in respiratory product development at Merck in Summit, NJ. He has authored and co-authored over 10 publications.
Dr. Oscar Liu is currently a Director of Preclinical Development, Pharmaceutical Sciences and Clinical Supplies, Merck Research Laboratories, Merck Co. and Inc., Summit, NJ where his primary focus is on the development and analysis of respiratory products (MDIs, DPIs, Nasal sprays and nebulizers) spanning from preclinical stage to commercial supply stage. Prior to joining Schering-Plough in 2007, Oscar had worked at Pfi zer (Groton, NJ) and Par Pharmaceutical (Spring Valley, NY) where he focused on oral controlled release dosage form development (NDA and ANDA fi ling). Oscar received a Ph.D. degree in 1998 from Duke University and an M.S. degree from SUNY-Binghamton in 1995. Oscar serves on the USP Expert Committee (2010-2015) and serves as a reviewer for the National Science Foundation and scientifi c journals; he is also a member of Governing Board of Eastern Analytical Symposium.
Dr. Brent Donovan is an Executive Director leading the respiratory product development department within Merck Research Laboratories in Summit, NJ. He leads a team of scientists that are engaged in analytical and formulation development of dry powder inhalers, metered dose inhalers, and nasal spray products. His group has been working on developing novel methodologies for the characterization of inhalation products as well as supporting numerous projects in all phases CMC development. He received his Ph.D. in Physical Chemistry from the University of Michigan. Dr. Donovan has several published original research articles within chemistry and pharmaceutical sciences. He previously worked at Schering- Plough for nine years prior to the merger with Merck in 2009.
References
- West, C.; Lesellier, E., J. Chromatogr. A, 2008, 1191, 21.
- West, C.; Lesellier, E., Adv. Chromatogr., 2010, 48, 195.
- West, C.; Lesellier, E., J. Chromatogr. A, 2006, 1115, 233.
- West, C.; Lesellier, E., J. Chromatogr. A, 2006, 1110, 181.
- West, C.; Lesellier, E., J. Chromatogr. A, 2006, 1110, 200.
- West, C.; Lesellier, E., J. Chromatogr. A, 2008, 1203, 105.
- Strubinger, J.R.; Song, H.; Parcher, J.F., Anal. Chem. 1991, 63, 104.
- Poole, C.F., J. Chromatogr. A, 2012, 1250, 157.
- Ashraf-Khorassani, M.; Taylor, L.T., J. Sep. Sci. 2010, 33, 1682.
- Gyllenhaal, O.; Edstrom, L.; Persson, B.A., J. Chromatogr. A, 2006, 1134, 305.
- Pinkston, J.D.; Stanton, D.F.; Wen, D., J. Sep. Sci. 2004, 27, 115.
- Zheng, J.; Glass, T.; Taylor, L.T.; Pinkston, J.D., J. Chromatogr. A, 2005, 1090, 155.
- Zheng, J.; Taylor, L.T.; Pinkston, J.D., Chromatographia, 2006, 63, 267.
- Taylor, L.T., J. Chromatogr. A, 2012, 1250, 196.
- Berger, T.A. ACS Symp. Ser. 1992, 488, 132.
- Brunelli, C.; Zhao, Y.; Hanna-Brown, M.; Sandra, P., J. Chromatogr. A, 2008, 1185, 263.
- Wang, Z.; Zhang, H.; Liu, O.; Donovan, B., J. Chromatogr. A, 2011, 1218, 2311.
- Mukherjee, P., J. Pharm. Biomed. Anal. 2007, 43, 464.
- Simmons, B.; Jagota, N.; Stewart, J., J. Pharm. Biomed. Anal. 1995, 13, 59.
- Bari, V.; Dhorda, U.; Sundaresan, M., Talanta, 1997, 45, 297.
- Bhoir, I.; Raman, B.; Sundaresan, M.; Bhagwat, A., Anal. Chim. Acta. 1997, 354, 123.
- Patil, S.; Sundaresan, M.; Bhoir, I.; Bhagwat, A., Talanta, 1998, 47, 3.
- Gyllenhaal, O.; Karlsson, A., J. Biochem. Biophys. Meth. 2000, 43, 135.
- Patela, Y.; Dhorda, U.; Sundaresan, M., Talanta, 1998, 47, 625.
- Karlsson, L.; Gyllenhaal, O.; Karlsson, A.; Gottfries, J., J. Chromatogr. A 1996, 749, 193.
- Gyllenhaal, O., J. Pharm. Biomed. Anal. 2006, 40, 971.
- Gyllenhaal, O.; Hulthe, J., J. Pharm. Biomed. Anal. 2002, 29, 381.
- Gyllenhaal, O., J. Chromatogr. A 2004, 1042, 173.
- Mukherjee, P.; Cook, S., J. Pharm. Biomed. Anal. 2006, 41, 1287.