It seems self-evident today, but worth remembering, that the pharmaceutical industry exists on a foundation of trust. Patients or even doctors have no way to actually determine the strength, purity and quality of the medicines prescribed and taken. Everyone trusts that the label is accurate and the medicines are pure. This was not always the case and efforts to safeguard our medicine supply led directly to USP, FDA and the GMPs.
Recently we have been reminded of the critical nature microbial Quality control plays in safe medications as contaminated medicine shipped nationally from a compounding pharmacy has sickened hundreds. The New England Compounding Center (NECC) of Framingham, MA was responsible for the manufacture of preservativefree methylprednisolone acetate. This was an aseptically produced parenteral, delivered intrathecally (directly to the spinal column, bypassing most of the body’s defense mechanisms).
It is difficult to envision a more hazardous situation and the results have been disastrous. Three lots of this product have exposed over 20,000 individuals to risk of fungal meningitis, and by latest count (April 15, 2013 - http://www.cdc.gov/hai/outbreaks/meningitis-map.html) have resulted in infections in 733 patients and 53 deaths associated with these intrinsically contaminated medicines.
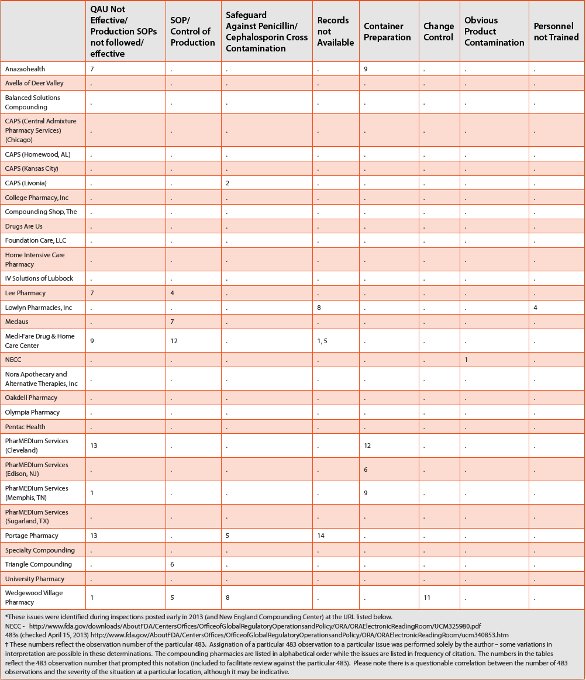
In response to this situation, FDA has embarked on an aggressive inspection schedule that resulted in multiple 483 findings in the beginning of 2013 (summarized in Table 1). Review of these 483 observations shows several common findings among the compounding pharmacies that received 483 observations during this time:
- Lack of procedures to prevent microbial contamination
- Problems with the Environmental Monitoring program
- Problems with batch release
- Lack of validation of the sterilization method
- Inadequate control/cleaning/qualification of critical equipment used in manufacture
- Issues with personnel gowning
- Expiry dating of manufactured medicines not supported by a stability study
- Issues with laboratory procedures or control of contract lab
- Issues with investigations
- Control of incoming raw materials and components
Table 1. Issues in Compounding Pharmacies Identifi ed by FDA 483 Observations*

Table 1. Issues in Compounding Pharmacies Identifi ed by FDA 483 Observation (cont.)

These (and others listed in Table 1) are basic GMP requirements, and described in 21 CFR 211 as well as many also being discussed in USP<797> Pharmaceutical Compounding – Sterile Preparations. For the moment, we will leave USP chapter<797> for discussion later in the article and focus for now on GMP. At a time when we are looking at compounding pharmacies that are functioning as pharmaceutical manufacturers, we need to look back at where pharma was and how we got here, and the genesis and development of the GMP as described in 21 CFR 210/211.
Table 1. Issues in Compounding Pharmacies Identifi ed by FDA 483 Observation (cont.)
FDA & GMP
A recent review [1] describes the development of FDA oversight of pharma. Originally the Drug Laboratory in the Bureau of Chemistry (US Department of Agriculture), it was created in 1906 through the Pure Food and Drugs Act. As an agency, however, it was toothless to aff ect the streams of fraudulent claims and questionable ingredients, (opium and morphine were very popular for their all-around curative properties), until the 1938 Food, Drug, and Cosmetic Act. This increased authority was direct response to the perceived need for a federal level control on medicines sparked by the production of a sulfonamide elixir using diethyl glycol in their oral product. This situation led to the death of 107 people. FDA was soon faced with another situation in 1941 with the release of sulfathiazole tablets containing phenobarbital as a contaminant. This incident led to the death or injury of over 300 people and served as the impetus for a drastic revision of manufacturing and quality control practices [2, 3]. The further expansion of FDA authority and GMP followed in a series of steps, all reactionary in response to a threat to the public health from manufacturers failing the public trust.
This failure was not always (or even usually) traceable to malfeasance. In fact, most of the cases seemed, in hindsight, to be due to ignorance on the part of the manufacturer as to the safety of his products (hence the requirement to document safety) or in the manufacturing process. A frequently cited example of this is the Cutter incident involving the polio vaccine [4]. The inactivation methodology for the polio virus was not well characterized before the process was approved for use by several manufacturers. One of these (Cutter) made small changes to the process that resulted in incomplete inactivation of the virus in the vaccine. At the height of the polio scare, this vaccine exposed about 200,000 people to live virus in the vaccine – 70,000 became ill and 200 were paralyzed with an additional 10 deaths.
The GMP has grown with FDA authority and in response to particular issues that have arisen. Validation of sterilization, change control, separation and product protection from cross-contamination with penicillin, tamper-resistant closures, etc. are all addressed in the GMP in response to an issue in the marketplace.
This, then, is the purpose of GMP – a preventive system to create processes and procedures that assure consistently high quality drugs, along with reactive programs to promptly detect and then prevent recurrences of problems. It is against this background that we examine at what is going on in compounding pharmacies.
Compounding Pharmacies and Contamination Issues
The basic assumption in compounding pharmacies is that the pharmacist is creating a specific formulation in response to a doctor’s script for a particular patient. This is a needed function in hospitals and street-corner pharmacies. However, it is not what is causing the problems we are reading about in the news. Some “pharmacies” are operating as pharmaceutical manufacturers, filling batches of thousands of units and then offering them for sale to doctors. These pharmacies, operating under widely varying standards frequently cite their lower costs over GMP manufactured medicines as a selling point. The potential issues this situation creates are obvious. It was precisely this type of situation that led to the generation of GMP.
This is not a small concern. According to a recent ISMP Medication Safety Alert [5]:
“The 2008 revision of the US Pharmacopeia (USP) Chapter<797> that left many facilities unable to meet the published standards for sterile compounding as well as the escalation in drug shortages have led to a steady increase in sterile compounding pharmacy services. A 2011 survey showed that 66% of hospital pharmacies outsource some portion of their sterile compounding. In some cases, pharmacists have purchased compounded products without full realization of the risks. An analysis of recent harmful cases of contaminated products from compounding pharmacies revealed breaches of USP <797>, unsafe staff behaviors, untrained and unskilled personnel, improper use of equipment, extended beyond use dating outside of manufacturer labeling without sufficient testing, and/or a lack of basic compounding skills involved in almost all cases. Outsourcing is also used as a cost-savings measure.”
We had previous warnings about the dangers of conditions in compounding pharmacies. As early as 1976, the widespread contamination of medications from hospital pharmacies was reported in the literature (9% contamination rate of P. aeruginosa alone [6]). By the mid-1990s FDA was investigating a number of pharmacies that were operating as manufacturing facilities in response to a number of contamination events [7]. This situation led directly to the generation of USP chapter <1206> Pharmacy Compounding Practices in 1996. This was a non-mandatory chapter in USP and was provided for informational purposes.
These problems remain. In 2005 CDC investigated a multi-state outbreak of Serratia marcescens that was traced back to intravenous magnesium sulfate from a compounding pharmacy [8]. This outbreak provided additional impetus for the revision of USP sterile compounding guidance to the current version of USP <797> Pharmaceutical Compounding – Sterile Preparations.
Even later, an industry group conducted a survey on pharmacy practices that described widespread belief of contamination in sterile medications prepared by the pharmacies [9]. While the risk may not be as severe for a hospital pharmacy, compounding a specific medication for immediate administration, in a manufacturing environment the longer storage times may result in microbial proliferation and product spoilage [10]. This concern is supported by current events. For example, FDA reported in 2007 that the Agency had reports of 200 adverse events at that time involving 71 compounded products since 1990 [5]. To put these numbers in context, it should be noted that adverse event reporting is rare for pharmacy products. This is because, in contrast to products that are subject to approved drug applications, there are no adverse event reporting requirements for drugs made by pharmacies. The general public has been made aware of some examples as well – a few of which are outlined in Table 2. As you review these events, please also note that many involve shipment across state lines, and sometimes involve extremely large batch sizes.
Table 2. Compounding Pharmacies and Microbial Contamination in the News*

USP and Compounding Pharmacies
The early history of USP parallels, in several respects, the recent contamination problems at NECC and the current state of regulation of compounding pharmacies. Early efforts (1790s-1810s) to create a pharmacopeia included the pharmacopeias of the College of Physicians (Philadelphia) and the Massachusetts pharmacopeia. However, not all of the newly-formed states adopted either of these pharmacopeias, which led to an effort to create a new pharmacopeia that enjoyed the support of all major medical societies and could serve as a “national” pharmacopeia. The first edition of this pharmacopeia was published in 1820. Throughout the 1800’s the compendia was periodically revised, with the participation of pharmacists. The 1906 Pure Food and Drug Act specifically cited USP and the National Formulary (NF) as enforceable standards. The 1938 amendment to the FD&C Act established FDA as the empowered enforcement agency, and again cited USP and NF for standards [11].
Having had a quick look at the history of USP, where a multitude of state-level compendia led to uneven standards, allow us jump to the current time. USP <797> is the recognized standard of practice for compounding pharmacies manufacturing sterile products in the USA. While this standard is a huge improvement over the previous “best practice”, it is far less stringent than the pharmaceutical GMP as described in 21 CFR 210/211. This is a point that must be remembered – USP <797> is clearly best practice among the top compounding pharmacies [12] but it is far less rigorous than the expectations of cGMP. This USP chapter’s guidance is appropriate for small pharmacies servicing specific prescriptions (its intended use), but is not adequate for large-scale production of pharmaceutical batches.
USP first published information on sterile compounding in 1995 in chapter <1206> “Sterile Products for Home Use” in USP 23 [13]. This was a general informational chapter on compounding pharmacy and not as effective as was originally hoped [14]. In respose, USP changed the informational chapter <1206> to the mandatory chapter <797> with the expectation that this change in status would allow enforcement of the provisions. It was also at this point that diff erent levels of “sterile” were incorporated into the chapter [12]. These levels of sterility included low, medium and high risk products based on compounding process, product characteristics and storage conditions. [15] This eff ort met with limited success. Voluntary compliance with USP and American Society of Hospital Pharmacies (ASHP) was low – estimated at 5.2% in a 2003 industry survey [12].
There were several “GMP”-like requirements that were new to the compounding pharmacy. Examples include the requirement for robust ISO Class 5 fi ll conditions, as well as the contamination control, facility, environmental monitoring, personnel gowning and training requirements.
However, these changes were not sufficient to address the continuing problems with compounding pharmacy Quality issues. In one case, for example, a “for cause” type of inspection ran into difficulty as the inspector objected to the lack of any written procedures. In reply, the pharmacist challenged the inspector to show any such requirement. This, and similar, experiences led to the revision of USP <797> in 2009 to incorporate several additional Quality controls [16].
It is interesting to note that at the time of this writing, there remain no uniform expectations for observance of USP <797> requirements and in fact the best estimate is that only 23 states currently require compliance with USP <797>[17]. A recent review article also highlighted the uneven training of pharmacists in the expectations of USP <797> [18]. Finally, we must remember that in comparison to cGMP, USP <797> Quality and safety standards are relatively lax as the expectation is that the medicine will be used immediately on a particular patient under a doctor’s specifi c direction. This is not the target market for the large compounding pharmacies like Massachusetts’ now bankrupt NECC.
Fda & Compounding pharmacies
Although FDA asserts jurisdiction over compounding pharmacies, as a practical matter, the stated policy of the Agency has been to restrict its attention to large-scale manufacturing in “compounding pharmacies” rather than the traditional creation of a specific formulation in response to a doctor’s prescription for a specific patient. As noted in a 2003 GAO report:
“FDA and others have also expressed concern about the potential for harm to the public health when drugs are manufactured and distributed in commercial amounts without FDA’s prior approval.
While FDA has stated that traditional drug compounding on a small scale in response to individual prescriptions is beneficial, FDA officials have voiced concern that some establishments with retail pharmacy licenses might be manufacturing new drugs under the guise of drug compounding in order to avoid FDCA requirements.” [19]
The concerns expressed by FDA were accurate, although this should be of little surprise given the history of FDA as the national guardian of safe medications. The situation, where a patchwork of state regulations and enforcement capabilities addresses compounding pharmacy practices, lends itself to uneven effectiveness. For example, in a 2003 testimony it was related that North Carolina has six inspectors for 2,000 pharmacies and claimed each was inspected at least every 18 months (this assertion works out to 1 inspector for every 333 pharmacies, with a work load of 19.5 pharmacy inspections with associated paperwork/ month – unless the inspector has field complaints to investigate, which take priority over inspections) [19].
This patchwork of regulation remains. The National Association of Boards of Pharmacy (NABP) relates that currently: “…at least 23 states require compliance with USP <797>… and seven additional boards indicate that they have rules that include some or most of the USP Chapter 797 standards. Three boards of pharmacy have such regulations pending, and another has regulations under consideration. In addition, Hawaii considers compliance with USP Chapter 797 a standard of practice, and the South Carolina Department of Labor, Licensing, and Regulation – Board of Pharmacy indicates that they have publically instructed licensees that it considers compliance with USP Chapter 797 to be appropriate professional practice and that it will consider serious deviation to be grounds for discipline.” [17]
As was described above, USP has been working to develop enforceable guidance for compounding pharmacies in chaptersand[12] but as of the time of this writing, state boards of pharmacy have yet to consistently include these expectations in their local standards [17]. Even in states that do include them, it is unclear whether the states have the resources to enforce the regulations [20]. This concern is supported by data. In a recent series of unannounced inspections by the Massachusetts’ Board of Pharmacy in the wake of the NECC scandal [21], only 4 of 40 compounding pharmacies met expected standards [22]. Eleven pharmacies were issued immediate cease and desist orders.
In a revealing letter to FDA, the American Society of Health-System Pharmacists (ASHP) argues against unfettered FDA oversight of large pharmacies arguing that they are a necessary part of American pharmaceutical service and should not be impeded [23]. Their position is stated as:
“ASHP has long recognized that hospitals may also enlist the help of qualified compounding pharmacies for some compounded preparations for several reasons. For example, they may not have necessary equipment or facilities to prepare some high-risk preparations, or they may face medication shortages for commercial products that can only be replicated by a compounding pharmacy. The Society’s policy position on compounding (excerpted) is as follows:
To affirm that extemporaneous compounding of medications, when done to meet immediate or anticipatory patient needs, is part of the practice of pharmacy and is not manufacturing;
- To encourage pharmacists who compound medications to use only drug substances that have been manufactured in Food and Drug Administration-approved facilities and that meet official United States Pharmacopeia (USP) compendial requirements where those exist;
- To encourage unaccredited facilities where extemporaneous compounding of medications occurs to seek accreditation by a nationally credible accreditation body;
- To advocate the adoption, in all applicable state laws and regulations governing health care practice, of the intent of the requirements and the outcomes for patient safety as described in United States Pharmacopeia Chapter 797.”
In other words, large scale compounding is required because smaller facilities may not have the expertise to make sterile products, and GMP facilities may have shortages. ASHP encourages (but does not support requiring) its members to meet USP <797> standards. The letter continues later:
“A sterile compounding business entity that does not fill prescriptions for individual patients is not a pharmacy. Regulatory oversight of these entities should be dependent on the scope and scale of their operations, which may range from patient-specific small batches to large-scale production of commonly used drugs or formulations based on historical demand. The beyond use date (BUD) or shelf life these entities assign to final products as well as the risk level (low, medium, high) of the compounding activity are also factors.
ASHP believes that the FDA has limited authority to inspect large scale compounding entities since most are licensed and operating as pharmacies. We believe that FDA’s authority needs to be clarified or new authorities given to FDA to regulate compounding businesses that produce large amounts of compounded products, and sell those products to entities other than the end user. …
The Society believe [sic] that compounding service providers that operate at the scale and scope of manufacturers should be required to register with the FDA, share details about their operations with the Agency, and submit to routine inspections.” [23]
While this last paragraph is encouraging, it seems clear that the safety of the public demands a nationally directed enforcement of GMP on all large-scale pharmaceutical manufacturers, even the ones who have been classified as compounding pharmacies.
Conclusions
GMP may be, at times, difficult to maintain, and sometimes seem overly proscriptive, but it provides a common set of expectations for the establishment and maintenance of controls over product Quality that require care and attention. It is vastly superior to the dangers of unregulated pharmaceutical products. In an industry dependent on trust, manufacturers and the public both need come commonly accepted practices to guide production as well as someone to police the less educated and prepared manufacturer. This is a national, not a state, issue as the medicines are shipped nationwide where they are needed. We have to be able to have confidence in the strength, quality and safety of our medicines. GMP is the rulebook by which this confidence is encouraged. The dangers of unregulated (or underregulated) production of medicines for national distribution are obvious in the news.
Author Biography
Scott Sutton, Ph.D., is the Principal of Microbiology Network, Inc. (http:// www.microbiologynetwork.com), a company he started in 1996 as a means to encourage training and communications within the microbiological community. He is a recognized consultant and trainer with emphasis in GMP, investigations, Environmental Monitoring and contamination control (both Aseptic manufacturing and non-sterile production facilities) as well as microbiology laboratory audits and operations. The Microbiology Network supplies consulting, training, webinars and e-mail discussion groups. Dr. Sutton is an active author and speaker for the industry, supports PDA and has served with the USP Analytical Microbiology Committee of Experts since 1993. He may be reached at [email protected].
Acknowledgements
I want to thank Dennis Guilfoyle, Radhakrishna Tirumalai, Anthony Cundell, and others for their helpful review and commentary on this manuscript.
References
- Wechsler, J. 2006. FDA Marks 100 Years of Drug Manufacturing Oversight. Pharm Technol 30(9):26-31
- Immel, B. 2000. A Brief History of the GMPs. Pharm Technol. 25(9):44-52
- Swann, JP. 1999. The 1941 Sulfathiazole Disaster and the Birth of Good Manufacturing Practices. PDA J Pharm Sci Tech. 53(3):148-153
- Offit, PA 2005. The Cutter Incident: How America’s First Polio Vaccine Led to the Growing Vaccine Crisis Yale University Press. ISBN 0-300-10864-8
- ISMP. 2012. Sterile compounding tragedy is a symptom of a broken system on many levels. http://www.ismp.org/Newsletters/acutecare/showarticle.asp?id=34 accessed 2/11/13
- Baird, RM et al. 1976. Pseudomonas aeruginosa In Hospital Pharmacies. Brit Med J 1:511- 512.
- Okeke, C., Newton, D., et al. 2001. History and Background Information on USP’s Activities in Compounding Pharmacy Practices/. Pharm Forum 27(5):3169 – 3172.
- Sunenshine, R et al. 2007. A Multistate Outbreak of Serratia marcescens Bloodstream Infection Associated with Contaminated Intravenous Magnesium Sulfate from a Compounding Pharmacy. Clin Infect Dis 45:527-33.
- ISMP. 2013. Survey results: How hospitals are managing the preparation and purchase of high-risk compounded sterile preparations (CSPs). http://www.ismp.org/Newsletters/ acutecare/showarticle.asp?id=40 accessed 2/10/13
- Lolas, A. & J. Metcalfe. 2011. Evaluation of the Microbial Growth Potential of Pharmaceutical Drug Products and Quality by Design. PDA J Pharm Sci Tech 65(1):63-70.
- Anderson, L and GJ Higby. 1995. The Spirit of Voluntarism A Legacy of Commitment and Contribution – The United States Pharmacopeia 1820-1995 United States Pharmacopeial Convention. ISBN 0-913595-88-8
- Newton, DW and LA Trissel. 2004. A Primer on USP Chapter“Pharmaceutical Compounding – Sterile Preparations” and USP Process for Drug and Practice Standards. Intl J Pharm Compounding 8(4):251-263.
- USP 1995.“Sterile Products for Home Use” USP 23/NF18 United States Pharmacopeial Convention pp. 1963-1975
- ASHP. 2008. ASHP Discussion Guide for Compounding Sterile Preparations: Summary and Implementation of USP Chapter. http://www.ashp.org/s_ashp/docs/files/ discguide797-2008.pdf accessed 2/8/13
- USP. 2004.Pharmaceutical Compounding - Sterile Preparations. USP 27/NF22 United States Pharmacopeial Convention. pp. 2350-2370.
- USP. 2009.Pharmaceutical Compounding - Sterile Preparations. USP 32/NF27 United States Pharmacopeial Convention. pp. 318-354.
- NABP. 2013. State Boards and NABP Take Action to Clarify Regulations and Bolster State Regulatory Systems Specific to Compounding Pharmacies Newsletter: National Association of Boards of Pharmacy February 42(2):25-30 http://www.nabp.net/system/redactor_assets/ documents/156/Final_February_2013_NABP_Newsletter.pdf accessed 2/14/13
- McElhiney, LF. 2012. Misinterpretation of United States Pharmacopeia ChapterIntl J Pharm Compounding 16(1):6-10.
- GAO . 2003. Prescription Drugs – State and Federal Oversight of Drug Compounding by Pharmacies. GAO-04-195T http://www.gao.gov/assets/120/110456.pdf accessed 2/14/13
- Massachusetts Special Commission on the Oversight of Compounding Pharmacies. 2013. Recommendations on the Oversight of Compounding Pharmacies in the Commonwealth. http://www.mass.gov/eohhs/docs/dph/quality/boards/pharmacy/pharmacy-commissionreport. pdf accessed 2/12/13
- Massachusetts Department of Public Health. 2012. New England Compounding Center (NECC) Preliminary Investigation Findings. http://www.mass.gov/eohhs/docs/dph/quality/ boards/necc/necc-preliminary-report-10-23-2012.pdf accessed 2/10/13
- Massachusetts Department of Public Health. 2013. Department of Public Health Announces Update on Unannounced Pharmacy Inspections. http://www.mass.gov/eohhs/ gov/newsroom/press-releases/dph/update-on-unannounced-pharmacy-inspectionsannounced. html accessed 2/10/13.
- ASHP 2013. Letter from ASHP to FDA January 13, 2013 http://www.ashp.org/DocLibrary/ Advocacy/GAD/Comments-to-FDA-on-Framework-for-Pharmacy-Compounding.pdf