Introduction
High performance liquid chromatography (HPLC) is the premier analytical technique used in many pharmaceutical applications including potency/purity/performance assays, pharmacokinetics/ bioanalytical testing, purification, high-throughput screening (HTS), In Process Control (IPC) Monitoring and Quality Control (QC) testing [1-6]. The pharmaceutical industry is the major consumer segment of HPLC [7] and has been the primary driving force for higher throughput and performance. This article provides a brief review of significant developments in HPLC impacting pharmaceutical analysis in the last decade. Topics discussed are listed below and selected items are shown in a representative timeline in Figure 1.
- Instrumentation: Ultra-high pressure LC (UHPLC) going mainstream.
- Columns: Sub-2 μm, sub-3μm core-shell and hybrid particles; novel bonding chemistries; hydrophilic interaction chromatography (HILIC); immobilized polysaccharide chiral phases; columns for biomolecules and biopharmaceuticals.
- Others: Liquid chromatography – Mass Spectrometry (LC/ MS) - particularly High-Resolution MS (HRMS) or Hybrid MS; Charged Aerosol Detector (CAD); Automated Method Development Systems (AMDS).

Figure 1. Newer developments (selected) in HPLC impacting pharmaceutical analysis in the last decade.
Our goal is to provide the busy pharmaceutical scientist with a concise yet comprehensive update of important HPLC developments, with each topic supported by brief descriptions of practical benefits/applications, critical commentaries from a user’s perspective, and key references.
Ultra-high pressure Liquid Chromatography (UHPLC)
The “revolution” in ultra-high pressure LC (UHPLC) began in 1997 with the proof-of-concept study by Professor James Jorgenson [8], followed by the first commercial system introduced in 2004 [1, 9-11]. Today, the transformation from HPLC to UHPLC is mostly complete with all major manufacturers having some type of UHPLC offerings. Detailed reviews of UHPLC systems, columns and applications are available elsewhere [4, 10-11]. Fundamentals, benefits, potential issues and best practices of UHPLC in pharmaceutical analysis are well documented [4, 11-14].
Table 1 summarizes the prominent characteristics and benefits of UHPLC. Note that higher system pressures allow the use of columns packed with smaller particles (e.g., sub-2 μm) for faster analyses (Figure 2) or superior separations of complex samples (Figure 3). All UHPLC have low system dispersion (~10 to 20 μL for 4σ bandwidths) from the use of improved injectors, smaller ID tubing (< 0.005”) and smaller UV detector flow cells (0.5 – 2 μL) [1, 9-11]. The use of 2.1 - 3.0 mm ID columns packed with sub-2 μm or sub-3 μm particles is typical. Other important system characteristics include smaller system dwell volumes (0.1 – 0.3 mL) and faster detector response/data acquisition (>40 pt/s) for high-throughput applications.
Table 1. Prominent Characteristics and benefits of UHPLC

The benefit of faster analysis of UHPLC vs. conventional HPLC is illustrated in Figure 2 in a method transfer case study for a drug product. Using a geometrical scaling of column and operating parameters from HPLC to UHPLC [11, 14, 15], a reduction of analysis time up to tenfold with similar resolution, is not unusual. This benefit of “faster analysis with good resolution” provides the primary incentive for most users to consider the purchase of the more expensive UHPLC equipment.
Another important benefit of UHPLC is its superior separation of complex samples. This aspect is often overlooked and appears to be “under-reported” in the literature [17-18]. Peak capacities (PC) in the range of 400 to 1000 in a reasonable time span (~1h) have been demonstrated using UHPLC. Peak capacity is the number of peaks that can be resolved in the chromatogram with a resolution of 1.0; typically ~200 for conventional HPLC [1]. For the first time ever, UHPLC can offer more effective assays, in a single dimension, for complex pharmaceuticals, natural materials, and other difficult sample matrices [4, 11, 17-18]. Figure 3a and 3b show two applications (a plant extract and a protein tryptic digest) to illustrate the current capability of UHPLC in high-resolution separations with Pc >300.
Other benefits of UHPLC include substantial solvent savings (5-15 fold), increased mass sensitivity (3 – 10 fold) and precision performance for both retention times (2 - 3 fold) and peak areas (<0.1% RSD) [11, 14, 18]. Note that reports on UV detection sensitivity increase with UHPLC are often misleading because mass sensitivity (amount of analytes injected) is primarily related to column void volumes. UHPLC typically does not increase concentration sensitivity (the most desirable kind of sensitivity) because one does not expect that the use of small flow cells can actually increase signal/noise ratio unless extended pathlength fflow cells (e.g., 60 mm) are used. Potential issues such as viscous heating, baseline perturbation from pump blending, and method transfer have been described and can be instrument-specific [4, 11, 18]. In general, these technical issues are well understood and can be readily mitigated by judicious choice of system configuration (e.g., mixer volume). Nevertheless, the implementation of UHPLC in QC labs remains to be time-consuming due to training, compatibility to “validated” data systems, and other method transfer issues [4, 18].
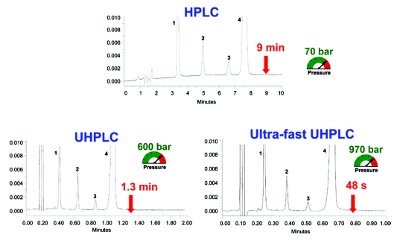
Figure 2. An example of a method transfer for a quality control assay of a commercial pharmaceutical formulation (Rapidocaïn) using geometrical scaling from conventional HPLC to UHPLC. Peak identifi cation: 1: methylparaben, 2: 2,6-dimethylaniline, 3 : propylparaben, 4 : lidocaïne. HPLC conditions: column RP18 150x4.6 mm, 5 μm, F=1 mL/min, Vinj=20 μL. UHPLC conditions: column RP18 50x2.1 mm, 1.7 μm, F=600 μL/min, Vinj=1.4 μL. Ultrafast UHPLC conditions: column RP18 50x2.1 mm, 1.7 μm, F=1000 μL/min, Vinj=1.4 μL [16].
One often hears arguments that UHPLC is perhaps not needed because other approaches (high-temperature LC, core-shell columns or 2-D LC) can be more cost-effective for enhancing speed or resolution. The reasoning is not truly valid since UHPLC can be used in combination with one or more of these approaches with superior results than those from conventional HPLC, as they are options rather than alternatives. Also, the term “UHPLC” may eventually go away in a few years since all newer HPLC will be UHPLC.
HpLC Column and stationary phase Developments
HPLC column is the heart of the chromatographic system. The pharmaceutical industry has been the primary driver for HPLC columns towards higher speed, resolution, and better peak shapes for basic analytes. In addition, QC laboratories have demanded improved column batch-to-batch reproducibility. From the 1970s to 1990s, there were steady quality improvements of the packing materials accompanied by a gradual reduction of “standard” particle sizes from 10 to 3 μm [1-3]. The introduction of high-purity type B silica materials (with low metallic content) in the late 1980s was a huge step and resulted in reduced silanol activity and substantial improvements in lot-to-lot consistency [21]. The use of high-purity silica is now the norm for all modern silica-based columns.
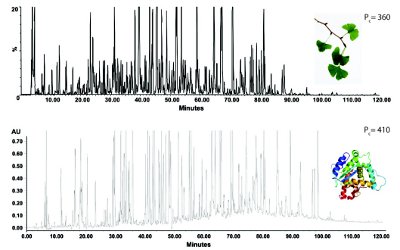
Figure 3. High resolution separation in UHPLC with C18 columns packed with sub-2 μm particles. (A) Chromatogram of a complex standardized extract of Ginkgo Biloba. Gradient from 5 to 40% acetonitrile in 120 min with T=30°C, Lcol=300 mm , F=0.2 mL/ min, ΔPmax=860 bar. Peak capacity was equal to 360 [19] (B) Chromatogram of a combined tryptic digests of three proteins. Gradient from 5 to 60% acetonitrile in 120 min with T=30°C, Lcol=40 cm, F=0.16 mL/min, ΔPmax=940 bar. Peak capacity was equal to 410 [20].
Recent developments in HPLC column technologies have been extensively reviewed [22]. Table 2 summarizes those with significant adoption rates by the industry for increasing productivity, stability, selectivity or retention (sub-2 μm, core-shell particles, hybrids, novel bonding chemistries, HILIC) or specialized applications (chiral compounds or biomolecules).
Table 2. Signifi cant HPLC column developments impacting pharmaceutical analysis

Sub-2 μm Particles
The use of very small particles for fast and efficient separations was predicted in 1956 by Van Deemter [23]. Typical particle size of packings has been decreasing in the past five decades cumulating to the use of sub-2 μm microparticulate silica in the early 2000s. As predicted, these particles (e.g., 1.7 μm) yield excellent efficiency performance (~280,000 plates/m or plate height of ~4 μm). However, columns packed with sub-2 μm particles generate high back pressures and are typically packed in a 2.1-mm ID format to reduce efficiency loss from viscous heating effects [4, 9, 14]. The system requirements for high pressures and low dispersion (to reduce extracolumn band broadening) were responsible for the current characteristics of modern UHPLC systems. Further reduction of particle size to less than 1.5 μm may be advantageous for even higher speed and performance. However, it must be accompanied by a substantial increase of system pressures and a reduction of column ID to capillary formats.
Core-shell Particles
The concept of fused-core or core-shell particles for reducing resistance to mass transfer was first described by Kirkland [24]. The first core-shell particles had these characteristics: 2.7 μm superficially porous silica materials with nonporous cores (1.7 μm) and porous shells (0.5 μm thick) [24-25]. These sub-3 μm particles appear to have similar efficiencies as the sub-2 μm fully porous materials but generate much lower pressure drops. The exceptional performance may be due to their shorter diffusion paths of the shells and/or the narrower distribution of the packings. Core-shell columns are rapidly gaining wide acceptance for fast separations (HTS, IPC and cleaning verification) and for biomolecules [25-27]. Myriad bonded phases and particle sizes (1.3, 1.7, 2.6, 2.7 and 5 μm) are currently available from an increasing number of manufacturers (6+). We fully expect these columns to be highly competitive in all applications to those from porous microparticulates [25].
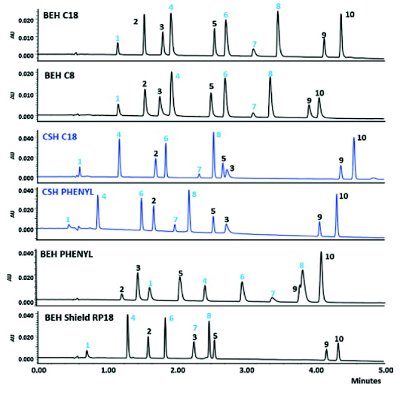
Figure 4. A comparison of separations of 10 commercial drugs with acidic, basic and neutral functional groups on BEH and CSH analytical columns using gradient elution with mobile phase containing acetonitrile and 0.1% formic acid. Peaks 1, 4, 6 and 8 were basic drugs. Reprinted from [31], with permission.
Hybrids
The concepts of hybrid particles with organic groups incorporated into the inorganic silica matrices were first described by Unger [28] in late 1970s, though the first commercial columns with methyl groups only became available in 1999 [29]. Bonded phases from these hybrids were demonstrated to have superior pH stability (from 1 to 12 with novel bonding chemistries vs. 2 to 8 for conventional silica with traditional monofunctional bonding), and lower silanophilic activity. A second-generation bridged ethylene hybrids (BEH) was introduced in 2005 and enjoyed immediate “mainstream” success, particularly for applications with high-pH mobile phase applications and UHPLC [9-10, 30].
Novel Bonding Chemistries
While the traditional monofunctional C18 silica-based bonded phase remains to be the main “staple” with perhaps the best batch-to-batch reproducibility [1, 3], newer bonding chemistries have brought benefits such as a wider pH stability range (from polyfunctional silane chemistry or silanes with isopropyl protective groups) and enhanced selectivity for difficult separations (polarembedded, phenyl, hexylphenyl, cyano, pentylfluorophenyl bonding) [1,3]. One recent innovation, termed charged surface hybrids (CSH) technology introduced in 2010, appears to have gained immediate acceptance for pharmaceutical analysis due to improved peak shapes of highly basic analytes under acidic, low ionic- strength mobile phases conditions (e.g., 0.1% formic acid) [31]. The proprietary technology involves the introduction of a low level of positive charges to the surface of the stationary phases. This is somewhat analogous to the addition of an amine additive such as triethylamine to the mobile phases in earlier days which is no longer acceptable in LC/MS due to ion suppression. Figure 4 illustrates the benefits of CSH particles in peak shape performance vs. comparable contemporary bonded phases [31]. Note that acidic compounds will have comparatively higher retention and increased peak tailing on CSH columns.
Hydrophilic Interaction Chromatography (HILIC)
The retention of many highly polar compounds is simply unachievable or problematic due to phase collapse (bonded phase “dewetting”) under reversed-phase LC (RPLC) conditions in mobile phases with low organic contents [1-3]. The HILIC mode, first developed by Alpert in 1990s [32], uses a hydrophilic stationary phase (silica, diol, cyano, amide, ztwitterionic) with an RPLC-like aqueous buffer and acetonitrile mobile phases, has enjoyed increasing popularity for the analysis of polar drugs, secondary drug metabolites, amino acids, peptides, neurotransmitters, oligosaccharides, carbohydrates, nucleotides, or nucleosides [33]. The actual retention mechanism in HILIC can be the “partitioning” of analyte molecules to the water layer adhering to the hydrophilic bonded groups. Other prominent benefits of HILIC include “orthogonal” selectivity to RPLC where sample preparations are compatible to both modes, higher electrospray ionization sensitivity for MS (5-15 fold), and lower operating pressures vs. RPLC.
Immobilized Polysaccharide Chiral Stationary Phases
Improved versions of the highly successful coated polysaccharides chiral stationary phases (CSPs) were available in the late 2000s. They brought similar versatility as the earlier coated CSPs but are more robust to aggressive solvents and can be used in normal phase, polarorganic and reversed-phase modes [5, 34].
Columns for Biomolecules
Wide-pore silica and polymeric packings first available in 1980s were effective for separations of large biomolecules [1, 35]. With the advent of recombinant proteins as biopharmaceuticals such as monoclonal antibodies (mAb), the need for detailed characterization by HPLC or capillary electrophoresis for QC purposes has become more pressing [36]. Recent developments of sub-2 μm microparticulates and coreshell wide-pore particles as well as several innovative ion-exchange and size exclusion phases have proved useful for separations of these large biologics [37].
High Resolution Mass Spectrometry (HRMS), Charged Aerosol Detection (CAD) and Automated Method Development Systems (AMDS)
Other high-impact developments in the past decade were in the HPLC detection and automation areas.
High Resolution Mass Spectrometry (HRMS)
The combination of HPLC with mass spectrometry (LC/MS) has been touted as the perfect analytical tool, combining the separation power of HPLC and the unsurpassed sensitivity/selectivity of MS. LC/MS is the preferred technique for identification of impurities and degradants,HTS in drug discovery, bioanalytical assays (LC/MS/MS for drugs and metabolites in biofl uids), and in-progress monitoring during process scale-up for the synthesis of drug substances [1-2, 5, 38]. LC/MS is becoming the standard platform technology for cleaning verification of highly-potent drugs [26] and determination of potential genotoxic impurities [2]. The last decade has seen the rapid evolution of HRMS (such as time-of-flight (TOF), OrbiTrap MS) and hybrid MS (such as Quadrupole-TOF or ion trap-OrbiTrap). The combination of HRMS with UHPLC and 2-D LC has enabled active research in metabonomics, proteomics, de nova protein sequencing, and characterization of biopharmaceuticals [5, 38]. Perhaps the most exciting opportunity for LC/MS lies ahead as a generic platform technology for the determination of disease biomarkers and clinical diagnostics in the emergent fi eld of personalized medicine.
Charged Aerosol Detection (CAD)
The lack of an ideal universal detector is often cited as a limitation of HPLC, though the UV/Vis detector comes fairly close for chromophoric compounds. The refractive index detector is not gradient compatible and does not have sufficient sensitivity [1-3]. Evaporative light scattering detection (ELSD) using nebulizer technology with laser light scattering detection is an option and is gradient compatible but has been recently surpassed by CAD (uses nebulizer with corona discharge detection), which offers better sensitivity (low ng) and improved linearity. CAD is becoming a mainstream detector for HTS in medicinal chemistry, reaction monitoring, and raw material/excipient testing [39].
Automated HPLC Method System (AMDS)
HPLC method development for complex mixtures is time-consuming due to the need to optimize many operating parameters (column dimension; type of bonded phase and mobile phase A and B (organic solvent/buffer type, pH, and ionic strength), gradient time and range, column temperature, and flow rate) [1, 5, 6]. A common example is the stability-indicating or purity assay of active pharmaceutical ingredients (API) in which all impurities and degradants must be separated for accurate quantitation by UV detection. Software or automated systems based on simulation, prediction, simplex optimization, and column/mobile phase screening have been available to facilitate HPLC method development for many years. Continued improvements have enhanced their capability and ease-of-use though they never appeared to be very popular [2, 6]. The latest market entry was an add-on software package compatible to two of the commonly used chromatography data systems. The software addresses the most time-consuming portion of the HPLC method development process (optimization) by automating method sequence from a user-defined design space using principles of Design of Experiments (DoE) and Quality by Design (QbD) [40-41]. This software can also perform statistical analysis and display optimum conditions after importing of the completed sequence results. Figure 5 illustrates the concepts and salient features of this software as implemented in our laboratory. Since concepts of DoE and QbD are well accepted, initial customer interest appeared to be high. AMDS is particularly useful to laboratories specializing in method development to support earlyphase drug development [2, 6]. For instance, to support process developments for API synthesis and drug product manufacturing from complex drug molecules with multiple chiral centers, as many as 10-20 HPLC methods (achiral and chiral methods for raw materials, starting materials, intermediates, final drug substances and drug products) are typically developed in quick succession.

Figure 5. Schematic diagram of an example instrument confi guration of an automated method development system using QbD software and a resulting graphics showing a design space with an operating region for optimum separations (white color).
Summary and Conclusions
In summary, HPLC remains a highly dynamic field with numerous innovations in instruments, column technologies, and approaches in recent years. Pharmaceutical scientists are early adopters and beneficiaries of these newer technologies for research, development and quality control. UHPLC is becoming the standard HPLC platform with rapid adoption by research & development, albeit slower implementation in QC labs. Newer column technologies allow faster and more efficient analysis of complex samples, chiral molecules and biomolecules. Finally, the rapid advancements of UHPLC and 2-D LC in combination with high-resolution MS have revolutionized life science research and promise to have even greater impact for clinical diagnostics. These developments are welcomed progress for the analytical chemist working in this rapidevolving world of drug development.
As a perspective for LC in the next years, some innovative works can be mentioned [42, 43]. As described by Professor Jim Jorgenson [42]: “Moving to still higher pressures (50,000 psi) will enable the use of smaller particles and/or longer columns, and yield faster and better separations. This will almost certainly require the use of sub-mm bore (capillary) columns due to issues with heat generation and dissipation. This won’t be easy, but the separation potential in terms of high speed with high resolution is enticing.” Alternatively, the use of extremely uniform packing of sub-micron silica particles in capillaries could be employed to generate plate heights well below 1 μm and impressive separations of protein variants [43]. These two research findings are unlikely to be ready for mainstream analytical work or QC very soon but the first results are encouraging and thus worth mentioning here.
Acknowledgements
The authors thank Dr. Dawen Kou, Dr. Tom Walter, and Richard Verseput for their suggestions and comments on this manuscript.
Author Biographies
Dr. Davy Guillarme holds a Ph.D. degree in analytical chemistry from the University of Lyon, France. He is senior lecturer at the University of Geneva in Switzerland. He authored 80+ journal articles related to pharmaceutical analysis and is editor of a recent book - UHPLC in Life Sciences, RSC, 2012. His expertise includes HPLC, UHPLC, HILIC, LC-MS, analysis of proteins and mAbs and SFC. He is an editorial advisory board member of Amer. Pharm. Rev. and LCGC.
Dr. Michael W. Dong is a Senior Scientist at Genentech, Small Molecule Analytical Chemistry and Quality Control, South San Francisco, CA. He was formerly Research Fellow at Purdue Pharma and Senior Staff Scientist at Applied Biosystems/Perkin-Elmer. He holds a Ph.D. in Analytical Chemistry from City University of New York, and a certificate in Biotechnology at U. C.Santa Cruz. He authored 80+ publications and 3 books including a bestseller in chromatography - Modern HPLC for Practicing Scientists, Wiley, 2006, and Handbook of Pharmaceutical Analysis by HPLC, Elsevier, 2005. He is an editorial advisory board member of Amer. Pharm. Rev. and LCGC.
References
- M.W. Dong, Modern HPLC for Practicing Scientists, Wiley, Hoboken, New Jersey, 2006.
- Y. V. Kazakevich and R. LoBrutto (Eds.), HPLC for Pharmaceutical Scientists, Wiley, Hoboken, New Jersey, 2007.
- L. R. Snyder, J.J. Kirkland, and J. W. Dolan, Introduction to Modern Liquid Chromatography, 3rd ed., Wiley, Hoboken, New Jersey , 2009.
- D. Guillarme, J-L Veuthey, and R. M Smith (Ed), UHPLC in Life Sciences, Royal Society of Chemistry Publishing, Cambridge, United Kingdom, 2012.
- S. Ahuja and M.W. Dong (Eds), Handbook of Pharmaceutical Analysis by HPLC, Elsevier/ Academic Press, Amsterdam, 2005.
- S. Ahuja and H. Rasmussen (Eds), HPLC Method Development for Pharmaceuticals, Elsevier/ Academic Press, Amsterdam, 2007.
- Market Analysis and Perspectives Report for Analytical and Life Science Instruments Industry, Strategic Directions Inc. Los Angeles, 2012.
- J. E. MacNair, K. C. Lewis and J.W. Jorgenson, Ultra High Pressure Reversed Phase Liquid Chromatography in Packed Capillary Columns, Anal. Chem. 69 (1997) 983-989.
- U. D. Neue, M. Kele, B. Bunner, A. Kromidas, T. Dourdeville, J. R. Mazzeo, E. S. Grumbach, S. Serpa, T. E. Wheat, P. Hong and M. Gilar, Ultra-Performance Liquid Chromatography, Technology and Applications, in Advances in Chromatogr. 48, CRC Press, Boca Raton, Florida, 2009, pp 99-143.
- K. J. Fountain and P. C. Iraneta, Instrumentation and columns for UHPLC separation. In UHPLC in Life Sciences, D. Guillarme, J-L Veuthey, and R. M Smith (ed), RSC Publishing, Cambridge, United Kingdom, 2012, pp. 283-311.
- M.W. Dong. Ultra-high-pressure LC in pharmaceutical analysis: Performance and practical issues. LC.GC 25(7), (2007), 656-666.
- N. Wu and A. M. Clausen, Fundamental and practical aspects of UPLC for fast separations, J. Sep. Sci. 30, (2007) 1167-1182.
- D.T.T. Nguyen, D. Guillarme, S. Rudaz, J.L. Veuthey. Fast analysis in liquid chromatography using small particle size and high pressure, J. Sep. Sci. 29 (2006) 1836-1848.
- D. Guillarme, J. Ruta, S. Rudaz, J.-L. Veuthey, New trends in fast and high-resolution liquid chromatography: a critical comparison of existing approaches. Anal. Bioanal. Chem. 397 (2010) 1069–1082.
- S. Fekete, I. Kohler, S. Rudaz, D. Guillarme, Importance of instrumentation for fast liquid chromatography in pharmaceutical analysis. J. Pharm. Biomed. Anal. http://dx.doi. org/10.1016/j.jpba.2013.03.012 (2013).
- D. Guillarme, D. Nguyen, S. Rudaz, J.L. Veuthey, Method transfer for fast liquid chromatography in pharmaceutical analysis: Application to short columns packed with small particles – Part I, isocratic separation, Eur. J. Pharm. Biopharm., 66 (2007) 475-482.
- D. Guillarme, E. Grata, G. Glauser, J-L. Wolfender, J-L. Veuthey and S. Rudaz, Some solutions to obtain very efficient separations in isocratic and gradient modes using small particles size and ultra-high pressure, J. Chromatogr. A 1216 (2009) 3232-3243.
- M.W. Dong, D. Guillarme, S. Fekete, R. Rangelova, J. Richards, D. Prudhomme, and N. P. Chetwyn, High-resolution separations of complex pharmaceuticals by UHPLC: Case studies and quality control implications, J. Pharm. Biomed. Anal., in preparation.
- E. Grata, D. Guillarme, G. Glauser, J. Boccard, P.A. Carrupt, J.L. Veuthey, S. Rudaz, J.L. Wolfender, Metabolite profiling of plant extracts by high temperature UHPLC-TOF/MS, J. Chromatogr. A, 1216 (2009) 5660-5668.
- J. Ruta, D. Guillarme, S. Rudaz, J.L. Veuthey, Comparison of columns packed with porous sub- 2μm and superficially porous sub-3μm particles for peptides analysis at ambient and high temperature, J. sep. sci., 33 (2010) 2465-2477.
- J. Kohler and J.J. Kirkland, Improved silica-based column packings for high-performance liquid chromatography, J. Chromatog. A 385 (1987) 125-150.
- T. L. Chester, Recent developments in HPLC stationary phases, Anal. Chem. 85 (2013) 579-589.
- J.J. van Deemter, F. J. Zuiderweg and A. Klinkenberg, Longitudinal diffusion and resistance to mass transfer as causes of non ideality in chromatography, Chem. Eng. Sci. 5 (1956) 271–289.
- J.J. Kirkland, T.J. Langlois, and J.J. DeStefano. Fused core particles for HPLC columns. American Laboratory 39 (2007) 18–21.
- S. Fekete, E. Oláh, J. Fekete, Fast liquid chromatography: The domination of core–shell and very fine particles. J. Chromatogr. A 1228 (2012) 57-71.
- M.W. Dong, E.X. Zhao, D.T. Yazzie, C. C. Gu, and J. D. Pellett. A Generic HPLC/UV Platform Method for Cleaning Verification, Amer. Pharm. Rev. 15(6) (2012)10-17.
- G. Guiochon and F. Gritti, Shell particles, trials, tribulations and triumphs. J. Chromatogr. A 1218 (2011) 1915-1938.
- K. K. Unger, Porous Silica: its properties and use as support in liquid chromatography, Elsevier, Amsterdam, 1979.
- U. Neue, T. H. Walter, B. A. Alden, Z. Jiang, R. P. Fisk, J.T. Cook, K. H. Glose, J. L. Carmody, J. M. Grassi, Y-f Cheng, Z. Lu and R. Crowley, Use of high performance LC packings from pH 1 to12, Amer. Lab. Nov. 1999.
- M.W. Dong, G. Miller, and R. Paul, MS-compatible ICH impurity analysis with a high-pH mobile phase: Advantages and pitfalls, J. Chromatog. A 987 (2003) 283-290.
- L. Novakova, H. Vlckova, P. Solich, Evaluation of new mixed-mode UHPLC stationary phases and the importance of stationary phase choice when using low ionic-strength mobile phase additives, Talanta 93 (2012) 99-105.
- A.J. Alpert, Hydrophilic-interaction chromatography for the separation of peptides, nucleic acids and other polar compounds. J. Chromatogr. A 499 (1990).177-196.
- P. Hemström and K. Irgum, Hydrophilic interaction chromatography. J. Sep. Sci. 29 (2006) 1784-1821.
- T. Zhang, P. Franco, D. Nguyen, R. Hamasaki, S. Miyamoto, A. Ohnishi, and T.Murakami, Complementary enantiorecognition patterns and specific method optimization aspects on immobilized polysaccharide-derived chiral stationary phases, J. Chromatogr. A 1269 (2012) 178-188.
- M.W. Dong, J.R. Gant and B. Larsen. Advances in Fast Reversed-phase Chromatography of Proteins, BioChromatogr. 4(1) (1989) 19-34.
- A. S. Rathore, Setting specifications for a biotech therapeutic product in the quality by design paradigm, Biopharm. International. 23(1) Jan. 2010.
- J. Jeong, T. Zhang, J. Zhang and Y-H Kao: UHPLC for Therapeutic Protein Characterization, Amer. Pharm. Rev, March, 2011.
- W.A. Korfmacher (Ed.), Mass Spectrometry for Drug Discovery and Drug Development Wiley, Hoboken, New Jersey, 2013.
- M. Swartz, M. Emanuele, A. Awad, and D. Hartley, Charged Aerosol Detection in Pharmaceutical Analysis: An Overview, LCGC, April 2009.
- Y.Li, G. T. Terfloth and A. S. Kord, A systematic approach to RP-HPLC method development in a pharmaceutical QbD environment, Amer. Pharm. Review, June 2009, 87.
- B. Debrus, D. Guillarme, and S. Rudaz, Improved quality-by-design compliant methodology for method development in reversed-phase liquid chromatography, J. Pharm. Biomed. Anal., in press.
- J. Jorgenson. Future trends in UHPLC. Presented at Pittcon 2013, Mar 19, 2013, Philadelphia.
- D.S. Malkin, B. Wei, A.J. Fogiel, S. L. Staats, M.J. Wirth, Sub-Micron Plate Heights for Capillaries Packed with Silica Colloidal Crystals, Anal. Chem., 82 (2010) 2175-2177.