Department of Formulation Sciences, Biopharmaceutical Development
Department of Formulation Sciences, Biopharmaceutical Development
Department of Formulation Sciences, Biopharmaceutical Development
Introduction
Biopharmaceuticals are routinely freeze-dried to improve product stability and, thereby, achieve acceptable commercial shelf life. However, freeze-drying is a unit operation coupled in formulation and process. While selection of excipients is primarily focused on improving product stability, for a freeze-dried product an additional consideration is the compatibility of the formulation with the freeze-drying process. The rational selection of excipients for freeze-dried product has been reviewed extensively [1-3]. This article will focus on freeze-drying process development after formulation selection. Understanding the impact of formulation and process parameters on drug product quality is critical to application of quality by design (QbD) principles, which aim to build quality within the process rather than monitoring it off-line at the end of the process. This article will lay out the approach and the significance for systematic formulation and process characterization for successfully designing and developing a robust freeze-drying process with acceptable product quality attributes. Also, an overview is provided of tools and techniques available to monitor and control the key freeze-drying process parameters during design, development, and scale-up. Lastly, the critical elements of QbD—Process Analytical Technology (PAT) and design space—are discussed for the primary drying step of the freeze-drying process.
Considerations for Freeze-Drying Process Design, Development and Scale-Up
The four steps outlined in Table 1 are key to successfully designing, developing, and scaling up the freeze-drying process.
Table 1. Considerations for Freeze-Drying Process Design, Development, and Scale-Up
Step 1: Pre-lyophilization Formulation Characterization
Thermal characterization of the formulation, using differential scanning calorimetry (DSC) and freeze-drying microscopy (FDM), is critical for defining the maximum allowable product temperature during the primary drying step of the freeze-drying process because drying above this temperature results in loss of cake structure. For an amorphous formulation matrix, the maximum allowable product temperature is Tg’ (the glass transition temperature of the maximally freeze-concentrated solution) or Tc (the collapse temperature). For a low protein concentration, the Tc is 1 to 2°C higher than the Tg’ and, hence, the maximum allowable product temperature during primary drying is usually set to Tg’. Typically drying above Tg’ results in cake collapse, which could impact product stability. However, for a high concentration protein formulation, visible collapse may not be observed for drying significantly above Tg’ [13-15], which could result in a shorter cycle time and high throughput. Further, having data to support that drying above Tg’ does not compromise product quality is useful, for processes where product temperature is maintained below Tg’, to support temperature and pressure excursions that are occasionally encountered on production-scale dryers. Figure 1 clearly shows that Tc is within 1 to 2°C of Tg’ up to a protein concentration of about 50 mg/mL. However, at a protein concentration greater than 50 mg/mL, the difference between Tg’ and Tc increases significantly, and at 100 mg/mL the difference is approximately 8°C. This difference is critical for defining the maximum allowable product temperature during primary drying because, for every 1°C increase in product temperature, drying time decreases by approximately 13%. Thus, a systematic thermal characterization of the formulation matrix is essential to design, develop, and optimize the freeze-drying cycle.
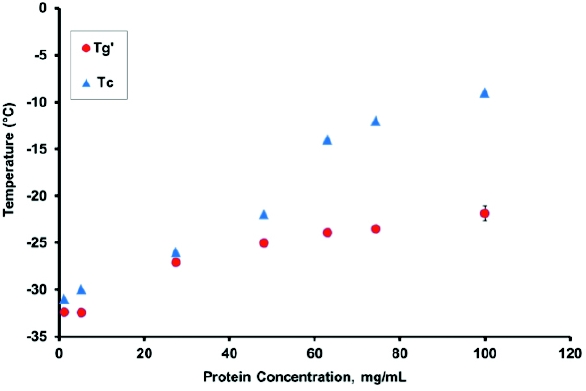 Figure 1. Tg’ and Tc as a Function of Protein Concentration in a Sucrose Containing Formulation Matrix
Figure 1. Tg’ and Tc as a Function of Protein Concentration in a Sucrose Containing Formulation MatrixStep 2: Freeze-Drying Cycle Development technology
The freeze-drying process is based on the fundamental principles of heat and mass transfer. The key objectives during freeze-drying process development are to:
- obtain stable and elegant product with minimal inter- and intra-batch heterogeneity,
- develop a process that is scaleable and readily transferable between lyophilizers,
- and minimize processing cost.
Defi ning and controlling the critical process parameters is essential to achieve these objectives. Several techniques that are widely used to monitor and control the critical process parameters are listed in Table 2. The application of some of these techniques is described in subsequent sections.
Table 2. PAT for Freeze-Drying Process Development

Step 3: process Characterization and robustness
Several mathematical models are available to model the freeze-drying process [4, 8, 10-12]. With these models, the number of experiments needed to assess the impact of process variables on product quality can be signifi cantly reduced. However, these theoretical models are only as good as the input parameters. A systematic characterization of the formulation and container closure system is required to obtain the input parameters for these models. Knowledge and experience gained by performing process characterization and robustness can be used to assess the impact on product quality of the temperature and pressure excursions typically encountered in a production scale freeze-dryer. However, the range of process parameters evaluated for each step should be based on the equipment capabilities and limitations to control these critical process parameters.
A comprehensive understanding of the formulation, process, equipment (freeze-dryer), and container closure system is essential to address four commonly encountered freeze-drying process development and scale-up challenges: freezing, edge vial effect, determining the end point of primary drying, and the effect of load.
Freezing
Ice nucleation is a random and stochastic process that results in freezing heterogeneity not only from batch-to-batch but also within a batch. Low ice nucleation temperature results in formation of smaller pores and, hence, higher product resistance and longer drying time. Because of the cleaner environment (class 100), the ice nucleation temperature in a production-scale freeze-dryer is typically much lower than that of a lab-scale freeze-dryer, making ice nucleation both a process development and a scale-up issue [16].
Annealing is commonly performed during the freezing step to not only crystallize the crystallizing excipients in the formulation matrix [17], but also to remove freezing heterogeneity and reduce the primary drying time [18, 19]. However, if annealing is applied, selection of the annealing temperature and time is critical. Annealing can potentially lead to product instability due to conformational change in protein structure or amorphous-amorphous phase separation [20, 21]. Hence annealing should be performed with caution.
Alternatively, commercial techniques can be applied to achieve spontaneous ice nucleation at the desired temperature via pressurization and depressurization of the product chamber with an inert gas [22]. This technique has shown potential for application even on a productionscale dryer [23]. Additionally, several other competing techniques [24, 25, 26] can allow controlling the ice nucleation temperature to address this process development and scale-up issue. However, application of many of these techniques on a production-scale dryer is challenging.
Edge Vial Effect [27]
Any vial that is not surrounded by six other vials is defi ned as an edge vial. During primary drying, edge vials receive additional heat transfer via radiation from the walls and door of the freeze-dryer, which are at a higher temperature than the shelf. This temperature diff erential results in higher product temperature and shorter primary drying time for edge vials compared to the rest of the batch (i.e., the center vials). During primary drying, it is critical to ensure that the product temperature remains below the maximum allowable limit not only for center vials but also for edge vials to maintain product quality across the batch. Also, the edge vial effect is more dominant in a lab-scale dryer due to the plexiglass door and surfaces with high emissivity. In a productionscale dryer, the door and surfaces are highly polished stainless steel with relatively low emissivity. These diff erences in dryer design need to be considered when scaling up the freeze-drying process to ensure that the product thermal profi le is not altered for both edge and center vials.
Determining the End Point of Primary Drying [28]
Insuffi cient primary drying time results in product collapse or melt back due to premature progression into secondary drying, whereas a prolonged primary drying time results in an unnecessarily long cycle time. Several techniques are now available to mark the end of primary drying; unfortunately, few can be easily applied to a productionscale dryer. The Pirani gauge is an inexpensive and reliable technique to determine the end of primary drying both on a laboratory and production scale dryer. During primary drying, the Pirani gauge reads approximately 60% higher than the capacitance manometer (CM) because the thermal conductivity of water vapor is approximately 1.6 times that of nitrogen. The Pirani pressure drops sharply toward the end of primary drying as the gas composition changes from mostly water vapor to mostly nitrogen [29]. A representative profi le of the Pirani pressure during primary drying is shown in Figure 2.
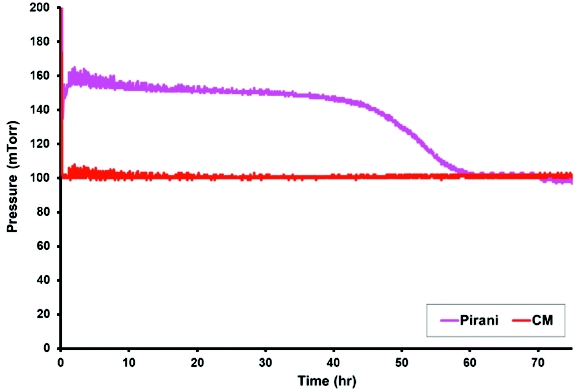 Figure 2. Typical Pirani Pressure Profile During Primary Drying
Figure 2. Typical Pirani Pressure Profile During Primary DryingEffect of Load [30]
As the load on the shelf increases, product temperature decreases and drying time increases. As shown in Figure 3, the primary drying time in diff erent freeze-dryers increases as the load increases. Dryer-X and Dryer-Y (lab-scale dryers) are similar in design and geometry and, therefore, the increase in primary drying time with batch size is similar. However, in Dryer-Z, which is a pilot-scale dryer, the same fraction of batch size takes longer to dry. Thus, understanding the effect of batch size on process performance and product quality is critical for defi ning the primary drying time.
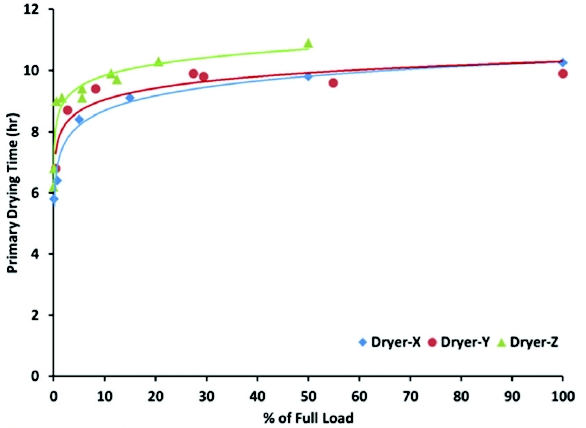 Figure 3. Effect of Load on Primary Drying Time
Figure 3. Effect of Load on Primary Drying TimeEquipment Characterization
Dryer capabilities and capacity during tech transfer and process scaleup are just as important as formulation and process characterization. The ice sublimation test is routinely performed to identify the limitations and capabilities of the freeze-dryer [31]. Figure 4 shows that the minimum achievable chamber pressure increases as the sublimation rate increases. Further, the trend of increasing chamber pressure with sublimation rate is dependent on dryer design and geometry. For example, Dryer-X and Dryer-Y, lab-scale dryers, show a similar trend, while Dryer-Z, a pilot-scale dryer, diff ers signifi cantly.
 Figure 4. Minimum Achievable Chamber Pressure as a Function of Sublimation Rate
Figure 4. Minimum Achievable Chamber Pressure as a Function of Sublimation RateThe limit (either due to choked fl ow or condenser overload) described by Figure 4 enables defi ning the Operational Space for a given dryer when designing and developing the freeze-drying process. Also, characterizing the ability of the freeze-dryer to control and maintain the shelf temperature (through shelf temperature mapping) and chamber pressure is critical for defi ning the practical range for process robustness. This systematic approach enables defi ning the Design Space and Control Space along with the Set Point for the primary drying step of the freeze-drying process as shown in Figure 5 [32]. Operating within the design space ensures that all product quality attributes will be met. The control space is based on the limitation and capabilities of the freeze-dryer to control chamber pressure and shelf temperature. The set point is selected within the control space such that the sublimation rate is fastest (i.e., shortest drying time), but with an appropriate safety margin to prevent a run-away process (i.e., loss of process control) due to choked fl ow or condenser overload.
 Figure 5. Design and Control Space, with Set Point, for the Primary Drying Step of the Freeze-Drying Process
Figure 5. Design and Control Space, with Set Point, for the Primary Drying Step of the Freeze-Drying ProcessStep 4: post-lyophilization product Characterization
Extensive characterization of the freeze-dried cake is critical for understanding the impact of formulation and process on drug product quality. Several techniques that are widely used to characterize both the freeze-dried cake and the diff erent physical states of the formulation matrix during freezing and drying are listed in Table 3. These techniques can be used to monitor the critical physical properties as the formulation matrix changes physical state from frozen to partially frozen and partially dried to dried and, fi nally, completely freeze-dried [33]. These physical properties have been shown to potentially impact product quality attributes such as residual water, reconstitution time, cake appearance, and physical stability.
Table 3. Techniques for Characterization of Frozen, Dried, and Freeze-Dried Product

Summary
Formulation and process development challenges need to be considered during early development to ensure product quality. Formulation characterization tools provide a macroscopic and microscopic comparative analysis to determine if any physical change occurred in the freeze-dried drug product. These tools are also valuable for comparative studies of freeze-dried products when changes are made to site, scale, process parameters, and drug product presentation. Lastly, PAT and design space are integral elements of QbD. PAT tools help confi rm that the lyophilization process is performing as expected, thereby assuring desired product quality.
Acknowledgements
The authors would like to acknowledge Nancy Craighead (Scientifi c Writing, MedImmune) for all her help with editing the manuscript.
Author Biographies
Dr. Sajal Manubhai Patel is currently working as a scientist at MedImmune in the department of Formulation Sciences. In his current role, Dr. Patel is responsible for formulation and drug product process development for both liquid and freeze-dried biologics. He is also in charge of developing strategy for freeze-dried drug product development and manufacturing. He received his M.S. in Chemical Engineering from University of Southern California and Ph.D. in Pharmaceutical Sciences from the University of Connecticut. His publications have focused on freeze drying process design and control. Dr. Patel’s research interests remain in this area and include application of QbD to the development and manufacturing of freeze-dried drug product.

Dr. Brian Lobo is a Senior Scientist in the department of Formulation Sciences at MedImmune. Dr. Lobo is responsible for formulation and drug product development of monoclonal antibodies and novel proteins as liquid and freeze-dried products. He has previously worked in formulation development at Amylin, Genentech and Merck. He received his B.S. in Pharmacy from Rutgers University and his Ph.D. in Pharmaceutical Chemistry from the University of Kansas. Dr. Lobo’s research interests include biophysical characterization and stabilization of proteins and peptides in liquid, frozen and freeze-dried formulations.

Dr. Ambarish Shah is a R&D Director in the Formulation Sciences Department at MedImmune. With over 15 years of leadership experience in small molecule and biopharmaceutical development at Wyeth, Human Genome Sciences, and MedImmune, he has supervised teams of up to 15 scientists from early development through post-commercial support and has led global teams and projects. He has led several high profile CMC investigations on commercial products leading to successful outcomes. He is a crossproject CMC Project team leader and a functional leader with expertise in biophysics, analytical technologies, lyophilization and drug delivery technologies. At MedImmune he is leading or actively supporting several technology initiatives, the Asia expansion initiatives, due diligences, alliance projects, and is a sitting member of several active committees and steering teams.
References
- Carpenter JF, Chang BS, Garzon-Rodriguez W, Randolph TW. Rational design of stable lyophilized protein formulations: theory and practice. Pharm Biotechnol. 2002;13:109-133.
- Carpenter JF, Pikal MJ, Chang BS, Randolph TW. Rational design of stable lyophilized protein formulations: some practical advice. Pharm Res. 1997;14:969-975.
- Pikal MJ. Freeze-drying of proteins: Part II: Formulation selection. Biopharm. 1990;3:26-30.
- Tang XC, Nail SL, Pikal MJ. Freeze-drying process design by manometric temperature measurement: design of a smart freeze-dryer. Pharm Res. 2005;22:685-700.
- Schneid SC, Gieseler H, Kessler WJ, Pikal MJ. Non-invasive product temperature determination during primary drying using tunable diode laser absorption spectroscopy. J Pharm Sci. 2009;98:3406-3418.
- Schneid SC, Gieseler H, Kessler WJ, Luthra SA, Pikal MJ. Optimization of the secondary drying step in freeze drying using TDLAS technology. AAPS PharmSciTech. 2011;12:379-387.
- Patel SM, Pikal M. Process analytical technologies (PAT) in freeze-drying of parenteral products. Pharm Dev Technol. 2009;14:567-587.
- Pikal MJ, Cardon S, Bhugra C, Jameel F, Rambhatla S, Mascarenhas WJ, et al. The nonsteady state modeling of freeze drying: in-process product temperature and moisture content mapping and pharmaceutical product quality applications. Pharm Dev Technol. 2005;10:17-32.
- Kuu WY, Nail SL. Rapid freeze-drying cycle optimization using computer programs developed based on heat and mass transfer models and facilitated by tunable diode laser absorption spectroscopy (TDLAS). J Pharm Sci. 2009;98:3469-3482.
- Giordano A, Barresi AA, Fissore D. On the use of mathematical models to build the design space for the primary drying phase of a pharmaceutical lyophilization process. J Pharm Sci. 2011;100:311- 324.
- Fissore D, Pisano R, Barresi AA. Advanced approach to build the design space for the primary drying of a pharmaceutical freeze-drying process. J Pharm Sci. 2011;100:4922-4933.
- Rasetto V, Marchisio DL, Fissore D, Barresi AA. On the use of a dual-scale model to improve understanding of a pharmaceutical freeze-drying process. J Pharm Sci. 2010;99:4337-4350.
- Colandene JD, Maldonado LM, Creagh AT, Vrettos JS, Goad KG, Spitznagel TM. 2007. Lyophilization cycle development for a high-concentration monoclonal antibody formulation lacking a crystalline bulking agent. J Pharm Sci 96(6):1598-608.
- Patel SM, Pansare S. 2012. Effect of Drying a High Concentration Mab at, above and below Tg’. PepTalk, San Deigo, CA.
- Mujat M, Greco K, Galbally-Kinney KL, Hammer DX, Ferguson RD, Iftimia N, Mulhall P, Sharma P, Pikal MJ, Kessler WJ. 2012. Optical coherence tomography-based freeze-drying microscopy. Biomed Opt Express 3(1):55-63.
- Rambhatla S, Ramot R, Bhugra C, Pikal MJ. Heat and mass transfer scale-up issues during freeze drying: II. Control and characterization of the degree of supercooling. AAPS PharmSciTech. 2004;5:e58.
- Li X, Nail SL. Kinetics of glycine crystallization during freezing of sucrose/glycine excipient systems. J Pharm Sci. 2005;94:625-631.
- Searles JA, Carpenter JF, Randolph TW. The ice nucleation temperature determines the primary drying rate of lyophilization for samples frozen on a temperature-controlled shelf. J Pharm Sci. 2001;90:860-871.
- Searles JA, Carpenter JF, Randolph TW. Annealing to optimize the primary drying rate, reduce freezing-induced drying rate heterogeneity, and determine T(g)’ in pharmaceutical lyophilization. J Pharm Sci. 2001;90:872-887.
- Heller MC, Carpenter JF, Randolph TW. Manipulation of lyophilization-induced phase separation: implications for pharmaceutical proteins. Biotechnol Prog. 1997;13:590-596.
- Lueckel B, Helk B, Bodmer D, Leuenberger H. Effects of formulation and process variables on the aggregation of freeze-dried interleukin-6 (IL-6) after lyophilization and on storage. Pharm Dev Technol. 1998;3:337-346.
- Konstantinidis AK, Kuu W, Otten L, Nail SL, Sever RR. Controlled nucleation in freeze-drying: effects on pore size in the dried product layer, mass transfer resistance, and primary drying rate. J Pharm Sci. 2011;100:3453-3470.
- Shon M. ControlLyoTM Nucleation On-Demand Technology Manufacturing Scale Implementation Case Study. PepTalk 2013;Palm Springs, CA.
- Geidobler R, Winter G. Controlled ice nucleation in the field of freeze-drying: Fundamentals and technology review. Eur J Pharm Biopharm. 2013.
- Ling M. Beyond Controlled Nucleation. PepTalk 2013, Palm Springs, CA.
- Brower J. Ice Fog incduced nucleation, CPPR Conference on Freeze-Drying of Pharamceuticals and Biologicals. . 2012.
- Rambhatla S, Pikal MJ. Heat and mass transfer scale-up issues during freeze-drying, I: atypical radiation and the edge vial effect. AAPS PharmSciTech. 2003;4:E14.
- Patel SM, Doen T, Pikal MJ. Determination of end point of primary drying in freeze-drying process control. AAPS PharmSciTech. 2010;11:73-84.
- Nail SL, Johnson W. Methodology for in-process determination of residual water in freeze-dried products. Dev Biol Stand. 1992;74:137-50; dicussion 150-1.
- Patel SM, Jameel F, Pikal MJ. The effect of dryer load on freeze drying process design. J Pharm Sci. 2010;99:4363-4379.
- Rambhatla S, Tchessalov S, Pikal MJ. Heat and mass transfer scale-up issues during freezedrying, III: control and characterization of dryer differences via operational qualification tests. AAPS PharmSciTech. 2006;7:E39.
- Patel SM, Pikal MJ. Lyophilization Process Design Space. J Pharm Sci. 2013.
- Liu J. Physical characterization of pharmaceutical formulations in frozen and freeze-dried solid states: techniques and applications in freeze-drying development. Pharm Dev Technol. 2006;11:3-28.