Abstract
The solubility of all drug substances is normally determined during the preformulation stage, and it is crucial to know whether the determined values represent genuine equilibrium solubilities (i.e., thermodynamic values) or whether they represent the values associated with a metastable condition (i.e., kinetic values). This review will provide an understanding of the distinction between thermodynamic and kinetic solubility, and how one can exceed the equilibrium solubility to yield a supersaturated solution. This requires one to determine if and when the substance is undergoing a physical change during the measurement period, and how any solubility values are to be assigned as reflecting either equilibrium solubility or metastable solubility.
The equilibrium solubility of a compound is defined as the maximum quantity of that substance which can be completely dissolved at a given temperature and pressure in a given amount of solvent, and is thermodynamically valid as long as a solid phase exists which is in equilibrium with the solution phase. The solubility properties of a drug substance are routinely determined as part of a preformulation program, since its dissolving tendencies will tend to govern the composition of its dosage form and its ultimate bioavailability.
Unfortunately, it is all too often that circumstances associated with either the substance or the dissolution medium lead to the formation of a supersaturated solution, where somehow an amount of solute exceeding the equilibrium solubility has become dissolved. Supersaturated solutions are thermodynamically metastable at best, and any such concentration value represents a kinetic solubility rather than an equilibrium solubility value intrinsic to the drug substance.
It is therefore necessary for an investigator to understand the distinction between thermodynamic and kinetic solubility, and to know when a particular measurement represents an equilibrium solubility value, or if the determined value simply represents some type of metastable condition. Since the most commonly encountered type of metastable solubility is observed in studies of the solubility of polymorphic forms of a drug substance, the principles involved will be illustrated through the analysis of particular systems.
Measurement of Solubility
Methods for the determination of solubility have been thoroughly reviewed [1-3], as have details of solubility issues specially related to crystal forms [4,5]. Most workers are familiar with the analytical method of solubility measurement, where the temperature of equilibration is fixed, and the concentration of solute in the saturated solution determined at equilibrium by an analytical procedure. For this method, the saturated solution is achieved by suitable contact between the undissolved solid and the solvent (at constant temperature) until equilibrium is reached. Equilibration is approached asymptotically at a rate that depends on the volume of the solvent, the surface area of the solid solute, and the nature and extent of the agitation. The method and time of equilibration should be established prior to the solubility determination by measuring the dissolved concentration of the solute as a function of time until no further change is noted, and analysis at times beyond the equilibration time is desirable to verify that a state of true equilibrium has been reached.
An equally valid analytical method for the determination of equilibrium solubility entails agitating a supersaturated solution until the concentration falls to a constant value. A plot of decreasing concentration as a function of time from a supersaturated solution should converge with the increasing plot of concentration obtained from an unsaturated solution, and when agreement is obtained between the two values, one may be certain that a reliable equilibrium solubility value has been measured.
Another procedure for determining equilibrium solubility is known as the synthetic method. Here, the composition of the solute-solvent system is fixed by the appropriate addition and mixing of the solute and solvent, and the temperature at which the solid solute just dissolves or just crystallizes is then carefully determined. For this method, either a weighed amount of the solute (or a definite amount of the solvent) is placed in a suitable vessel, and then the system is agitated at constant temperature. Known amounts of the solvent (or the solute) are added gradually until the solubility limit is reached, and appropriate checks must be carried out to ensure that the system is very close to equilibrium when the content or temperature of the system is recorded.
Solubility is ordinarily quite dependent on temperature, and an endothermic enthalpy of solution implies that the solubility will increase with increasing temperature. Conversely, an exothermic enthalpy of solution implies that the solubility will decrease with increasing temperature. Most solids (but not all) are characterized by endothermic enthalpies of solution, while most gases are characterized by exothermic enthalpies of solution. Plots of solubility against temperature are commonly used for characterizing pharmaceutical solids [6], and especially over a relatively narrow temperature range, a linear relationship may be obtained using either the van’t Hoffequation:
or the Hildebrand equation:
In equations (1) and (2), X2sat is the mole fraction solubility of the solid solute at an absolute temperature T, a is the apparent molar enthalpy of solution, b is the apparent molar entropy of solution, and c’ and c” are constants.
Thermodynamic Solubility: Systems Remaining Phase-stable during Performance of a Solubility Study
It is very clear that the validity of any solubility measurement requires that a given crystal phase must remain stable with respect to any phase conversion taking place during the determination of the equilibrium solubility. Typically, the X-ray powder diffraction (XRPD) pattern of the initial substance is compared to the XRPD pattern of the solid phase recovered after the equilibration process. These two patterns will be the same if the crystallographic form of the solute has remained unchanged during the process. If the existence of a solution-mediated phase transformation is demonstrated by a change in XRPD pattern as a result of the equilibration, then the measured solubility value would necessarily be that of the new solid phase of the solute and not that of the original substance.
For polymorphic systems that remain phase-stable during solubility measurements, their intrinsically different lattice energies are manifested in different enthalpies of fusion and different melting points. These differences yield different slopes and intercepts for the ideal solubility lines, and at a given temperature, the polymorph with the lower solubility must be the more stable form.
Polymorphic systems are classified into two types, depending on the possibility of reversible phase transformations. Enantiotropy describes the situation where one polymorph is the most stable phase within a definite range of temperature and pressure, and the other form is the most stable phase over a different range of temperature and pressure. The phase interconversion of enantiotropic systems is reversible, and so the polymorphic identity of the substance will be governed by its environment. The temperature dependence of the solubility of an enantiotropic polymorphic pair is illustrated in Figure 1, and the temperature at which the solubility curves intersect is termed the transition point of the system.
 Figure 1. Temperature dependence of two enantiotropic polymorphs of a hypothetical substance, where Form-A is characterized by an enthalpy of fusion equal to 19.0 kJ/mol and a melting point of 165°C and Form-B is characterized by an enthalpy of fusion equal to 14.0 kJ/mol and a melting point of 175°C.
Figure 1. Temperature dependence of two enantiotropic polymorphs of a hypothetical substance, where Form-A is characterized by an enthalpy of fusion equal to 19.0 kJ/mol and a melting point of 165°C and Form-B is characterized by an enthalpy of fusion equal to 14.0 kJ/mol and a melting point of 175°C.Other systems exist where only one polymorph is stable at all temperatures below the melting point. Since all other polymorphic forms have no region of stability anywhere on a pressure-temperature diagram, they must therefore be metastable with respect to the stable form. Polymorphic systems of this type are said to exhibit monotropy, and the polymorph having the lowest free energy and solubility at any given temperature will necessarily be the most thermodynamically stable form. The temperature dependence of the solubility of a monotropic polymorphic pair is illustrated in Figure 2, where it is to be noted that there is no temperature at which the solubility curves intersect.
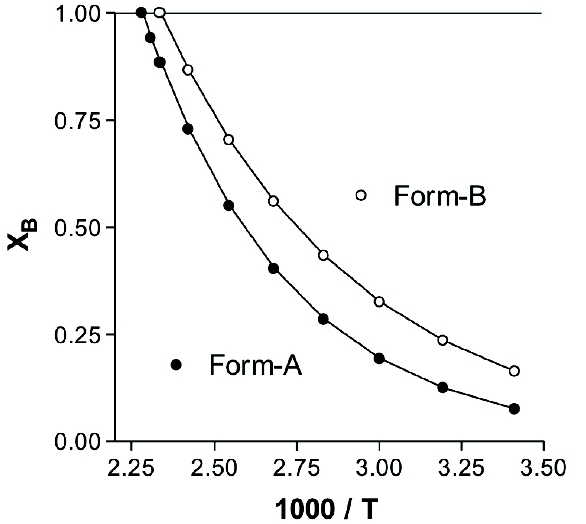 Figure 2. Temperature dependence of two monotropic polymorphs of a hypothetical substance, where Form-A is characterized by an enthalpy of fusion equal to 19.0 kJ/mol and a melting point of 165°C and Form-B is characterized by an enthalpy of fusion equal to 14.0 kJ/mol and a melting point of 155°C.
Figure 2. Temperature dependence of two monotropic polymorphs of a hypothetical substance, where Form-A is characterized by an enthalpy of fusion equal to 19.0 kJ/mol and a melting point of 165°C and Form-B is characterized by an enthalpy of fusion equal to 14.0 kJ/mol and a melting point of 155°C.Phenylbutazone is known to exist in at least five different polymorphic forms which are easily distinguished by their physical properties [7]. Form-A was found to exhibit the lowest solubility and intrinsic dissolution rate in phosphate buffer, demonstrating it to be the thermodynamically stable polymorphic form at room temperature. Since no change in crystal form took place during the dissolution or solubility studies, the stability order of the polymorphic forms was established as Form-A (solubility = 4.80 mg/mL) > Form-B (solubility = 5.10 mg/mL) ≈ Form-D (solubility = 5.15 mg/mL) > Form-E (solubility = 5.35 mg/mL) > Form-C (solubility = 5.90 mg/mL).
Kinetic Solubility: Systems Not Remaining Phase-stable during Performance of a Solubility Study
Because only one polymorphic form of a substance can be the most thermodynamically stable form under a defined set of environmental conditions, one frequently observes that a particular crystal form undergoes a solution-mediated phase transformation during the time required to establish an equilibrium solubility [8]. Conversions of a metastable phase into a more stable phase may include processes such as transformation of one polymorphic phase into another, solvation of an anhydrous phase, desolvation of a solvate phase, transformation of an amorphous phase into a crystalline anhydrate or solvate phase, and degradation of a crystalline anhydrate or solvate phase to an amorphous phase.
Since determinations of the solubility of solid materials are often made by suspending an excess of the compound in a dissolution medium, transformation of a metastable phase to a more stable one will yield a determination of the solubility of only the stable phase. The metastable phase must have a higher free energy than the thermodynamically more stable phase, and so will undergo a phase transformation once the activation energy barrier is overcome. A corollary of this is that if the barrier toward phase transformation cannot be overcome, then the solubility of the metastable crystal form can be measured using conventional means.
For example, two of the polymorphic forms of sulfathiazole were found to be unstable when contacted with water, and only Form-III exhibited constant dissolution rates during its characterization [9]. The initial dissolution rates of Form-I and Form-II were measurably different, but as the experiment progressed, the dissolved concentration values converged on the solubility of Form-III. The equilibrium solubility of either Form-I or Form-II could not be determined, as these converted into Form-III before equilibrium could be achieved.
One can take advantage of a solution-mediated phase transformation to determine which crystal form is the more thermodynamically stable phase. If a system moving toward equilibrium is seeded with a different crystal form during the experiment, and one observes a significant decrease in measured concentration, then the phase of the seed crystal represents the more stable polymorph or solvatomorph. Such phase conversion is also often observed during the dissolution of an amorphous substance, and the time dependence of the dissolution curve will resemble that shown in Figure 3. In this study, the initially high solubility of the amorphous form was found to drastically decrease after approximately 10 minutes owing to the spontaneous self-nucleation of the crystalline form.
 Figure 3. Dissolution curve of the stable crystal form of a developmental drug substance (open squares) and that of the amorphous form of the same substance (open circles). The solution of the amorphous form was found to undergo self-nucleation of the crystal form after approximately 10 minutes elapsed time.
Figure 3. Dissolution curve of the stable crystal form of a developmental drug substance (open squares) and that of the amorphous form of the same substance (open circles). The solution of the amorphous form was found to undergo self-nucleation of the crystal form after approximately 10 minutes elapsed time.Solution-mediated phase transformations are strongly affected by the composition of the dissolution medium, with this phenomenon being especially important for transitions between hydrate and anhydrate phases. For example, it was found that water activity was the major thermodynamic factor determining the nature of the solid form of ampicillin which crystallized from methanol/water mixtures [10]. In such systems, the control of solvent activity can be exploited to minimize solution-mediated phase transformations, enabling one to obtain true equilibrium solubility values through the use of appropriate solvent combinations.
Several indirect methods have been proposed to determine the solubility of metastable polymorphs. Milosovish [11] deduced the relative solubilities of metastable and stable polymorphs based on the measurement of intrinsic dissolution rates, while Ghosh and Grant [12] proposed an extrapolation technique to determine the solubility of a crystalline solid undergoing a solution-mediate phase.
The time evolution of light scattering from aqueous suspensions of anhydrous theophylline has been used as a means to evaluate its solubility, and also to study its phase transformation into its monohydrate solvatomorph [13].
Conclusions
Determination of the equilibrium solubility of a phase-stable crystal form is relatively straightforward, and accurate values can be obtained without too much difficulty. The same methodologies can be used to determine the equilibrium solubility of alternate crystal form as long as the energy barrier between the metastable polymorph and the stable polymorph is sufficiently high so that interconversion during the lifetime of the measurement does not take place. If, however, the free energy difference between the polymorphs (which is the driving force behind the phase transformation) is sufficient to overcome the activation energy barrier, then the solubility of the metastable polymorph can only be estimated through the use of more advanced techniques.
References
- D.J.W. Grant and T. Higuchi. Solubility Behavior of Organic Compounds, John Wiley and Sons: New York, 1990.
- S.H. Yalkowsky and S. Banerjee. Aqueous Solubility Methods of Estimation for Organic Compounds, Marcel Dekker: New York, 1992.
- P. Augustins and M.E. Brewster. Solvent Systems and Their Selection in Pharmaceutics and Biopharmaceutics. Springer-AAPS Press: Arlington, VA, 2007.
- H.G. Brittain. Solubility Methods for the Characterization of New Crystal Forms, Chapter 3.7, in Preformulation in Solid Dosage Form Development, M.C. Adeyeye and H.G. Brittain, Eds. Informa Healthcare Press: New York, 2008, pp 323-346.
- H.G. Brittain; D.J.W. Grant; and Paul B. Myrdal, Effects of Polymorphism and Solid- State Solvation on Solubility and Dissolution Rate, Chapter 12, in Polymorphism in Pharmaceutical Solids, 2nd edition, H.G. Brittain, Ed., Informa Press: New York, 2009, pp 436-480.
- D.J.W. Grant; M. Mehdizadeh; A.H.-L. Chow; and J.E. Fairbrother. Int. J. Pharm. 1984; 18: 25-38.
- M.D. Tuladhar; J.E. Carless; and M.P. Summers. J. Pharm. Pharmacol. 1983; 35: 208-214.
- H.G. Brittain. Solid-State Phase Transformations, Chapter 13 in Polymorphism in Pharmaceutical Solids, 2nd edition, H.G. Brittain, Ed., Informa Press: New York, 2009, pp 481-509.
- M. Lagas and C.F. Lerk. Int. J. Pharm. 1981; 8: 11-24.
- H. Zhu and D.J.W. Grant. Int. J. Pharm. 1996; 139: 33-43.
- G. Milosovish. J. Pharm. Sci. 1964; 53: 484-487.
- S. Ghosh and D.J.W. Grant. Int. J. Pharm. 1995; 114: 185-196.
- H.G. Brittain. Langmuir 1996; 12: 601-604.
Author Biography
Harry G. Brittain is the Institute Director of the Center for Pharmaceutical Physics, a research and consulting institute he established in 1999 for the study of substances having pharmaceutical interest. His research interests include all areas of pharmaceutical physics and physical pharmacy, including preformulation, formulation design, and product characterization. Dr. Brittain has authored over 320 research publications and book chapters, and edited the monographs Physical Characterization of Pharmaceutical Solids, Polymorphism in Pharmaceutical Solids (first and second editions), Spectroscopy of Pharmaceutical Solids, and Preformulation in Solid Dosage Form Development. He is the Editor-In-Chief of the book series Profiles of Drug Substances, Excipients, and Related Technology.