Creating and delivering safe and effective products that reliably meet our customers’ needs, to improve patients’ health and quality of life, is central to health care industry objectives.1 Advances in our understanding of a range of diseases have resulted in new therapies, and new ways of administering those therapies. These include targeted application of a combination of drugs and/or biological products with medical devices to minimize adverse events, while improving treatment response and user experiences.
Product development begins with gaining a clear understanding of the customer, and then within that context, defining user needs, intended uses and use environments for a given product. Achieving safe, effective, high quality products that meet customer needs necessitates robust product and process design. Combination products, containing two or more different types of regulated constituents (Figure 12-5), bring added complexity in the product development process and life cycle management.
 Figure 1. Combination products: Contain two or more diff erent types of regulated constituents
Figure 1. Combination products: Contain two or more diff erent types of regulated constituentsIn this article, we compare and contrast the drug and device development processes, and then integration of the two for developing combination products. We then discuss some of the challenges associated with combination product manufacturing.
During the development of combination products, factors that impact the constituent parts of the combination products should be considered. For example, for drug device combination products, one must distinguish between a basic drug container closure system and containers that are also devices. The key difference is generally whether the article is designed to deliver the drug it contains, or merely to hold it.6 A drug container closure system includes both primary and secondary packaging components, if the latter provides additional protection to the drug product. If a container closure system simply holds the drug, it is subject to the drug current Good Manufacturing Practices (cGMPs) as a container or closure. If it both holds the drug and delivers the drug, it may be considered a medical device, too. In this case, the drug cGMPs apply, and the Device Quality System Regulations (QSRs) may also apply. Metered dose inhalers and piston syringes are examples of elements of container closure systems that are device constituent parts.
Under 21 CFR Part 4 (Final Rule),7,8 the United States Food & Drug Administration (FDA) codifies a “Streamlined Approach” for compliance with combination product cGMPs. This “Streamlined Approach” leverages the fact that there are many common elements between drug, device, biologic and cell tissue therapy regulations, and identifies which elements of these regulations are uniquely interpreted based on constituent parts of the combination product. It helps to ensure requirements are appropriately implemented, while avoiding unnecessary redundancy in cGMP operating systems. For a manufacturer of a combination product operating under a device-based quality system, key considerations for demonstrating compliance with provisions of drug requirements (21 CFR part 211) are identified. For a manufacturer of a combination product produced under drugbased (21 CFR Parts 210/211) quality systems, key considerations for demonstrating compliance with provisions of the device regulations (21 CFR Part 820) are identified (Figure 2 and Table 1). For example, if a firm traditionally manufactures pharmaceuticals, they are likely operating under a drug-based quality system. If they are manufacturing a drug pre-filled syringe, in addition to the drugbased quality system quality system, they are responsible for 21 CFR 820.20 (Management Responsibility), 21 CFR 820.30 (Design Controls), 21 CFR 820.50 (Purchasing Controls) and 21 CFR 820.100 (CAPA) as interpreted under the Device Quality System Regulations.
 Figure 2. Combination Product cGMPs for Co-Packaged Combination Products and Single Entity Combination Products *
Figure 2. Combination Product cGMPs for Co-Packaged Combination Products and Single Entity Combination Products *Table 1. “Streamlined Approach”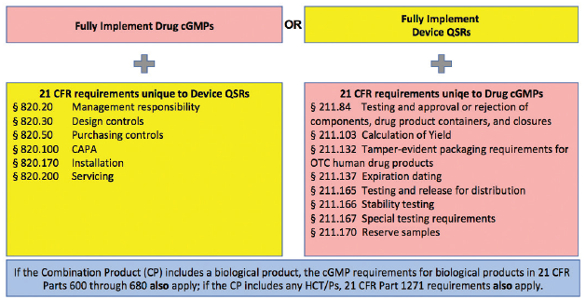
Now let’s review the combination product development process in the context of combination products regulations, comparing and contrasting the drug and device development processes, and then integration of the two. Generally, product development can be described in multiple stages from early research, defining user needs, then translating those user needs into product design requirements, doing iterative design and development, developing manufacturing processes and test methods, conducting verification and validation, all culminating in product launch and lifecycle management. This is the case whether developing a drug, a medical device, or a combination product. Quality by Design (QbD) and Design Controls are approaches for drug and device product and process development, respectively, that focus on improving chances of product development success by proactively addressing risks throughout requirements definition, development and lifecycle management.
 Figure 3. Typical Drug Development Process: Quality by Design
Figure 3. Typical Drug Development Process: Quality by DesignDrug development typically is done following Quality by Design (QbD) principles (Figure 3: Typical Drug Development Process).1 QbD is a “systematic approach applied during drug development that begins with predefined objectives and emphasizes product and process understanding and process control, based on sound science and quality risk management.”9 QbD principles are applied throughout the Chemistry, Manufacturing and Controls (CMC) process, facilitating robust product outcomes. According to Yu et al.10 the goals of pharmaceutical QbD are four-fold:
- Achieve clinically relevant product quality specifications;
- Enhance product and process knowledge, design, understanding and control, driving robust process capability, reduced product variation and defects;
- Increase efficiencies throughout product development and manufacturing; and
- Improved root cause analysis and post approval change control.
In the drug development process (Table 2: Typical Drug Development Clinical Studies),1 the pre-clinical discovery process begins with efforts to gain new insights into disease processes that can be translated into products to slow, stop or reverse the effects of a disease.1 During feasibility, perhaps thousands of molecular compounds are vetted, with the intention of identifying a few promising compounds, and ultimately selecting a new molecular entity (NME) that has the desired therapeutic benefit(s). In building further understanding of the potential benefits and mechanisms of action, research focuses on identifying side effects (toxicity) using both in vitro and in vivo methods. Through progressive stages of clinical development, detailed information is generated on dosing and toxicity levels to inform decisions on whether the drug is safe and effective for its intended use in humans.
Table 2. Typical Drug Development Clinical Studies 
Device development is done following Design Controls (Figure 4: Typical Device Development Process).1 Similar to QbD, Design Controls is a systematic approach applied to development that begins with predefined objectives and emphasizes product and process understanding and process control, based on sound science and quality risk management. Design Controls begins by defining user needs and desires, and customer-centered-insights. These user needs and intended uses are translated into device product attributes and functional requirements (Design Inputs). Through iterative design-build-test cycles, where device design, bench testing, and if needed, animal testing/clinical trials are used (Table 3: Device Development Clinical Studies),1 Design Inputs are translated into product and process specifications (Design Outputs). Device clinical trials are designed to support a “reasonable assurance of safety and effectiveness” for the marketing application. Conflicting requirements must be considered. For example, in design of a combination product, such as an auto-injector for self-administration by a rheumatoid arthritis patient, one would need to consider specifications for actuation force. One wants that force low enough so that the patient is readily able to self-actuate the device to administer the drug, but not so low as to allow the device to pre-maturely actuate. Such conflicting requirements may be identified proactively using tools such as Harm/Hazards Analysis, user- and/or designFailure Modes and Effects Analysis (FMEA).8 Formative human factors studies may be useful in refining such specifications over the course of development. Design Verification and Validation are conducted to ensure outputs meet input requirements and that the medical product meets user needs and intended uses. Summative human factors studies are an important part of combination product design validation activities.11, 12
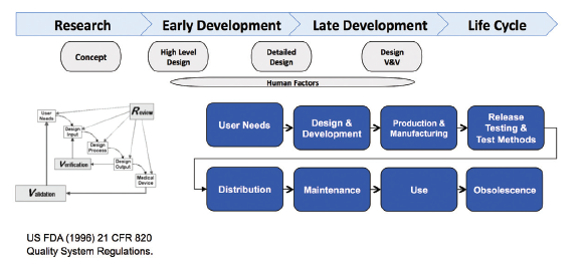 Figure 4. Typical Device Development Process: Design Controls
Figure 4. Typical Device Development Process: Design ControlsTable 3. Typical Device Development Clinical Trials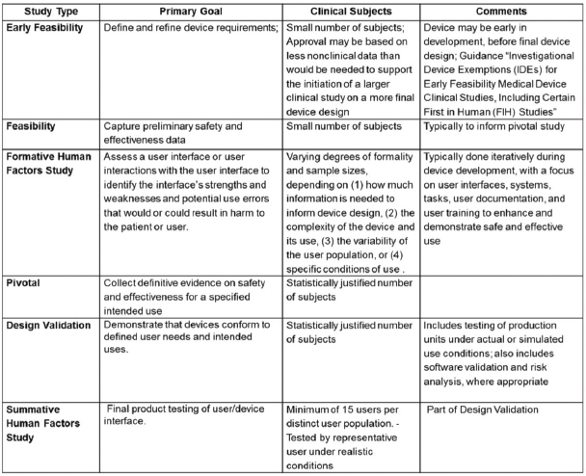
Proactive risk management is an essential aspect of both QbD (ICH Q9) and Design Controls (ISO 14971).13 For each, risk management is linked throughout the quality management system in order to enable risk based decisions throughout the product life cycle.
Combination products development necessitates consideration of the drug and device functionality and interactions during research and throughout development and life cycle management (US FDA 21 CFR Part 4). Figure 5 illustrates the integrated development process for a combination product with a drug-primary mode of action. Drug and device constituent development processes run in parallel, but have key intersections. Of particular importance is the use of commercially representative product, or its equivalent, for final combination product design verification and design validation activities. “Difficulties can occur when the development process for a device constituent part does not align with the overall development process for the combination product. The FDA recommends that Phase 3 clinical studies be conducted using the ‘to be commercialized’ combination product (i.e., the proposed commercial equivalent device used with the proposed commercial drug formulation).”8 The intent is to ensure that the functionality, safety and efficacy of the scaled-up combination product (drug and device and final integrated product) have been vetted, verified and validated to proactively identify and mitigate performance risks).
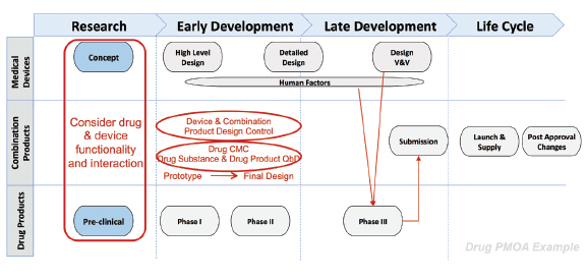 Figure 5. An Integrated Drug and Device Development Process for Combination Products
Figure 5. An Integrated Drug and Device Development Process for Combination Products
As indicated earlier, there are many common elements between Drug cGMPs and QSRs. Figure 6 (QbD and Design Controls Similarities)1 illustrates parallels between traditional drug development QbD and device development Design Controls. Executing Design Control or QbD development processes ensures diligence in product development and enables robust project management. While there are nuanced differences in requirements that must be met to demonstrate compliance, recognizing similarities may help an organization align their drug and development processes for successful outcomes.
 Figure 6. Drug Development (QbD) and Device Development (Design Controls) Similarities.
Figure 6. Drug Development (QbD) and Device Development (Design Controls) Similarities.Combination Products Purchasing Controls
When considering integrated drug and device development for combination products, purchasing controls are vital, as many combination products include purchased materials, components or device assemblies. Under 21 CFR 820.50, FDA states that “each manufacturer shall establish and maintain procedures to ensure that all purchased or otherwise received product and services conform to specified requirements.” This applies both to purchased products and to services. For drug Primary Mode of Action, it is not unusual, for example, to outsource the design of the device constituent, or to utilize an existing device platform. This changes the “Design” process, because it does not start from rough drawings, but rather, starts with selection. Selecting components and device constituents to be incorporated into the combination product is the device design. Purchasing controls assure the rigor required during technology evaluation, selection and life cycle management. Purchasing controls assure the rigor required during technology evaluation, selection and life cycle management. Implementation of purchasing controls as part of combination product design supports a proactive, rather than a reactive, regulatory strategy. Supplier Quality Agreements are a critical tool for suppliers and customers, in which they agree on proactive notification, review and approval of changes. Suppliers should also ensure that their upstream suppliers have adequate change control programs in place.
Combination Products Manufacturing
With the backdrop of the integrated drug and device development process, let’s turn our focus to combination products manufacturing. Manufacturing process development and validation begin with the drug substance and extend through to the drug product for clinical and commercial phases. For purposes of combination products, we’ll explore the drug product manufacturing at a typical fill-finish site. The drug product serves as a drug constituent part of the combination product. From a design controls perspective, for a drug primary mode of action, the drug product can be considered as a design input.
Let’s first take a look prior to the fill-finish process of a typical pre-filled syringe manufacturing process (Figure 7: Typical Compounding Process). Prior to fill-finish, drug substance is shipped and stored frozen to avoid microbial growth. Sterile equipment is used throughout the process. The formulated bulk drug substance is thawed and pooled in a class 10,000 cleanroom environment and mixed to achieve homogeneity. To ensure that the required bioburden thresholds are met, an in-process test is conducted pre-filtration. Media is transferred from the pooling and mixing vessel through a filter, into a storage vessel. The media is now ready for use to fill the syringes.
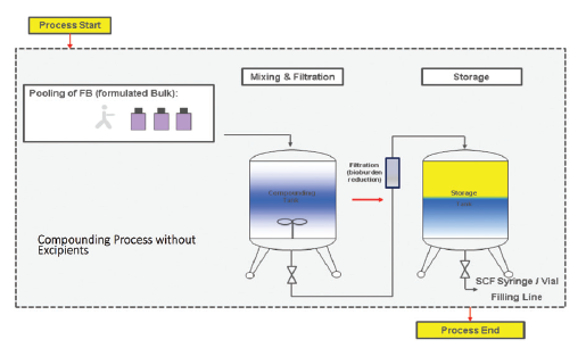 Figure 7. Typical Compounding Process
Figure 7. Typical Compounding ProcessPrior to filling, syringes and stoppers are prepared (Figure 8: Typical Filling Process). (1) Syringes are delivered in a sterile-ready-to-fill state (sterile from the supplier). Stoppers are supplied as pre-washed, in ready-to-sterilize bags. Off -line autoclave sterilization of stoppers is performed. (2) The tub that will be used to hold the syringes is decontaminated using e-beam sterilization. The production line is also decontaminated prior to any use. At this point we are ready to start the filling process. As an extra precaution, in order to ensure bioburden is kept to an absolute minimum, the media is sterile filtered using micro-porous filters. (3) Filling begins in a Class 100 clean room environment. Filling of syringes is done with an in-line weight check to ensure fill volume accuracy. Following filling, stoppering is done. Stoppering is controlled with an off -line check. (4) Filled syringes are placed in tubs and transferred outside the Class 100 clean room back into a Class 10,000 clean room environment. (5) Tubs each have unique identification codes for traceability. The tubs are then transported (6 & 7 to cold storage in a cold storage chamber. Unique identifier labeling on the syringes (color code) and optical inspection for defects (cracks, particles) are subsequently conducted. The pre-filled syringe is now ready for any required subsequent assembly and/or packaging.
 Figure 8. Typical Filling Process
Figure 8. Typical Filling ProcessWith the typical filling process as context, let’s step back and look at manufacturing challenges. Main challenges include: (a) Drug Substance Formulation, (b) Flexible Facility Designs, (c) Drug Product Manufacturing, (d) Stability Considerations and (e) Manufacturing Logistical Considerations. These are described below:
- Drug Substance Formulation: When developing a combination product, potential interactions between the drug, its excipients, and the device constituents must be carefully identified and controlled. The design control and risk management process must take this into account.
- Flexible Facility Designs: Shared facilities are often the standard for fill-finish operations. This requires diligence to avoid any cross-contamination issues. Changeover from one product to another has to be planned and efficiently executed to minimize down-time. A robust cleaning validation program is critical to ensure there is no cross-contamination. The isolator (Figure 8) must be completely cleared of the previous batch, including any support materials (like empty stopper bags, tools or other equipment); The entire isolator must be decontaminated, and tanks and small parts must be cleaned and sterilized. Then all preparations for the next batch must be addressed. Procedures and checklists are helpful to ensure robust changeover.
- As far as Drug Product Manufacturing, clean room requirements must be maintained according to industry guidelines. Effective filling equipment sterilization is critical. The quality of utilities, such as those for waterfor-injection, gas for filtration, and air particulates, both viable and non-viable, must be maintained. Monitoring of in-process critical parameters is essential throughout the manufacturing process. Risk-based methods should be used to determine the particulate monitoring strategy, depending on, for example, factors such as the type of asceptic process and its proximity to filling. Placement of active or passive monitoring settle plates in defined locations is essential, as is making sure there are enough of them. It is not possible to 100% check your product for sterility, as it requires destructive testing.
Aseptic Processing Simulation is used to help ensure controls developed will provide adequate process control in use. Simulation is done using a nutrient medium in the process. As part of the assessment, pre-filled syringe units are filled with a nutrient medium, incubated and tested for microbial growth. This simulation approach ensures that no colony forming units (CFU’s), indicating lack of sterility, are formed. Examples of routine and simulated interventions that are used as part of the simulation process are listed in Table 4. For a new filling process, the process simulation challenge must be repeated, with representative batch sizes, to support validation. Periodically, simulations are repeated to ensure the process remains in control. Prior to significant changes to the actual process, such as installation of new or modified critical components (e.g., dosing system, process set-up, filling unit, or line speed), one should repeat the validation.
Table 4. Drug Product Manufacturing: Aseptic Processing Simulations-Examples of Routine Interventions and Simulations 
Based on the nature of the drug constituent part, holding times and conditions are a critical factor impacting product quality. Holding times during the manufacturing process must be wellunderstood, controlled, validated and maintained during drug product filling, and device assembly and packaging. For the most part, sterile drug product is stored under refrigerated conditions (2-8°C). Device assembly and packaging are conducted at ambient conditions. Given sensitivity of drug product to temperature during any processing, careful monitoring and control of time outside refrigeration is needed. Production planning must account for hold times given varied batch sizes. For example, in manual assembly processes, only partial lots are processed at a time, to ensure cold chain requriements are met. Holding times are based on available stability data. Adherence to holding time is essential for batch release decisions.
It is essential that critical process parameters be monitored throughout manufacturing. In-process testing is used to evaluate product throughout the process, end-to-end, identify undesirable trends relative to established control limits, and to make appropriate adjustments to ensure the process remains within validated conditions. In process controls may be in-line, closed loop controls, such as automated check-weigh stations for 100% assessments enabling real-time adjustment to volume dosed if required. Alternatively, in-process controls may be done to isolate reject parts, sorting them from the process if control limits are exceeded. This may be done, for example, for attribute testing, e.g., for missing labels or missing parts. Alternatively, manual off -line testing may be conducted based on samples drawn from the line, with appropriate reactions. This is higher risk as it typically requires longer test result lead times, and can even result in rejects of portions of a batch.
Assembly and labeling are further challenges. The assembly process and tertiary packaging (e.g., folding box, tray, Instructions for Use, label) must consider container closure integrity requirements to maintain product sterility. Design of the assembly process, whether it be executed manually, semi-automated or fullly-automated, is critical to ensure product quality. Human interfaces are the most impactful aspects of the processing particularly for manual processes. Operators require clear working instructions and specifications. In the semi-automated assembly process, the process consists of both manual and automated steps, with manual loading and removal of product from equipment. Increasing automation requires robust validation to ensure proper functioning of system. Regardless of level of automation, Critical Process Parameters must be characterized and qualified.
Stability Considerations: Normal storage and excursions must both be considered. For biological products, refrigeration is typically required (e.g., 2-8°C). Shelf life of the product must be determined through stability studies of the drug product, the device constituent, and interactions between the two. Drug degradation may be a factor and device constituent functionality must both be assessed. Impacts can be due to interaction effects or due to storage conditions. For example, cold storage may make the device constituent more brittle, or could impact its functionality, while the same cold storage prolongs drug product expiration dating. The shelf life of the product is the shorter of the constituent parts. Temperature excursions must also be characterized during stability studies to determine what might be acceptable if temperature ranges are exceeded during transport. During transport, the storage conditions are monitored and each package has a unique temperature recording device to ensure storage conditions were acceptable.
d. Manufacturing Logistical Considerations: On a day-today basis, there are additional challenges, in part due to the global nature of supply chains, and differing product configuration or labeling requirements country by country. This results in a high number of packaging material combinations. Proposed changes must be carefully evaluated and implementation effectively managed, to be consistent with local country requirements.
Conclusions:
In summary, integrated drug and device development for combination products must consider drug and device functionality and interactions. Quality by Design (QbD) and Design Controls are approaches for drug and device product and process development, respectively, incorporating risk-based strategies to effectively support such integrated development, and to assure safe, effective combination products that meet user needs. They both focus on improving chances of product development success by proactively addressing risks throughout requirements definition, development and lifecycle management. Often constituents, components or services are outsourced for combination products. Outsourcing design, or utilizing an existing device platform, changes the “Design” process, necessitating robust application of Purchasing Controls to assure rigor during technology evaluation, selection and life cycle management.
Manufacturing combination products has its unique challenges. Manufacturing challenges that must be addressed span the gamut of drug formulation, facility designs, drug product manufacturing, stability considerations, and logistical challenges. Understanding and controlling interactions of drugs and devices throughout the process end-to-end is important in addressing these challenges.
References
- Neadle, Susan (editors: Bills, E. and Mastrangelo, S.) (2016). “Risk Management Considerations and Strategies in Product Development” in Lifecycle Risk Management for Healthcare Products: From Research Through Disposal. Davis Healthcare International Publishers.
- US FDA (1996) 21 CFR 820 Quality System Regulations.
- US FDA 21 CFR 210 Current Good Manufacturing Practice in Manufacturing, Processing Packaging, or Holding of Drugs; General.
- US FDA (1978) 21 CFR 211 Current Good Manufacturing Practice for Finished Pharmaceuticals.
- US FDA 21 CFR 600-680 Biological Products Standards
- US FDA (January 2017) Current Good Manufacturing Practice Requirements for Combination Products FINAL GUIDANCE
- US FDA (2013) Federal Register, Vol. 78, No. 14, January 22, 2013, Department of Health and Human Resources, Food and Drug Administration, Docket No. FDA-2009-N-0435, Current Good Manufacturing Practice Requirements for Combination Products, Final Rule.
- AAMI TIR48:2015. Quality Management System. (QMS) Recommendations on the. Application of the U.S. FDA’s CGMP. Final Rule on Combination Products. American. National. Standard.
- International Conference on Harmonization (ICH) (2009a)/US Department of Health and Human Services, Food and Drug Administration. Guidance for Industry: Q8 (R2) Pharmaceutical Development.
- Yu, L. X. et al. (2014) Understanding Pharmaceutical Quality by Design. The AAPS Journal, 2014 Jul; 16 (4):771-783. Epub 2014 May 23.
- BS EN 62366-1:2015 Part 1: Application of Usability Engineering to Medical Devices
- US FDA (February 2016) Applying Human Factors and Usability Engineering to Medical Devices: Guidance for Industry and Food and Drug Administration Staff
- EN ISO 14971 (2012) Medical devices – application of risk management to medical devices (CEN).
Author Biographies

Susan Neadle is currently Senior Director, Quality Engineering & Design-to-Value, and Head, Combination Products Quality at Janssen Pharmaceuticals. In this global role, she leads her organization to build proactive quality into the entire value chain, to prevent defects during lifecycle management, and to enable new and evolving business models and portfolio complexity. She successfully led Janssen’s Combination Products initiative to define the business model, develop and integrate crossfunctional capabilities, ensuring compliance while maintaining business growth momentum. Susan was recognized with the Janssen Supply Chain Inclusion Award for Leadership- Building a World Class Combination Products Center of Excellence. Susan also leads a cross-functional Designto-Value Core Team to develop and embed an array of new and improved processes across R&D, Supply Chain and Commercial organizations, driving greater customer value and improved business performance. She is active in Working Groups in International Society of Pharmaceutical Engineers, AdvaMed, Parenteral Drug Association, and Combination Products Coalition, and serves as a speaker, trainer, moderator and panelist at multiple venues. She recently authored the chapter “Risk Management Considerations and Strategies in Product Development” in the book Lifecycle Risk Management for Healthcare Products: From Research Through Disposal.
Over her extensive career, Susan has held positions of increasing responsibility in R&D and Quality Assurance in Pharmaceutical, Medical Device and Consumer segments. Susan is a Design Excellence Black Belt, and has applied that expertise while playing integral leadership roles in R&D product & process development of several new products. She has had responsibility for new and marketed products that span a diverse portfolio of Pharmaceuticals, Biologics, Devices and Combination Products. She holds multiple patents, and has received multiple awards recognizing her technical leadership and business impacts, including J&J’s highest award for innovation, the Johnson Medal, and the J&J Patent of Commercial Significance Award.
Susan holds a B.S. in Biology/Chemistry from State University of New York at Albany, an M.S. in Polymer Science & Engineering from University of Massachusetts at Amherst, and is a Fellow in the American Academy of Optometry.
Imre Váradi is a member of the Quality Assurance leadership team for Janssen Supply Chain in Schaffhausen, Switzerland. In his role, he is responsible for the site Quality Assurance aspects of combination products during both market introduction and lifecycle management. He also has end-to-end accountability for fill-finish of new parenteral drug products in the precommercialization phase.
Imre earned his M.S. in Chemical Engineering. During his career at Johnson & Johnson he has worked in analytical R&D, quality control and quality assurance positions. Prior to joining Johnson & Johnson he held Quality positions at Merck and Alcon. In these positions, he worked in analytical development, quality management, quality assurance and supplier management.