The views and opinions expressed in this article are only of the authors and do not necessarily reflect the views or policies of the FDA.
Introduction

In the pharmaceutical industry, particle characterization of powder materials has become one of the crucial aspects in drug product development and quality control of solid oral dosage forms. The particle size distribution (PSD) of the drug substance may have significant effects on final drug product performance (e.g., dissolution, bioavailability, content uniformity, stability, etc.). Furthermore, the PSDs of both drug substance and excipients can affect drug product manufacturability (e.g., flowability, blend uniformity, compactibility, etc.), which, ultimately, can impact safety, efficacy, and quality of the drug product. Many publications have shown that the PSDs of pharmaceutical powders have profound influence on almost every step of manufacturing processes for solid oral dosage forms, including pre-mixing/mixing, granulation, drying, milling, blending, coating, encapsulation, and compression [1-3]. Therefore, the impact of particle sizes of pharmaceutical powders on drug product manufacturability and performance should be evaluated at different pharmaceutical development phases for each specific drug application [4]. Once these particle size effects have been determined in the final development phase, the target PSDs of pharmaceutical powders can be selected and the appropriate particle size specifications should be established for control of drug product quality and ensuring manufacturing consistency. In this paper, we will discuss several important aspects of the particle size specification for solid oral dosage forms from a regulatory perspective. Discussion will include the relevance of a particle size specification as a part of the critical material attributes of formulation components and product intermediates as well as key considerations in the establishment of a particle size specification.
When is a Particle Size Specification Needed?
For the drug substance, the International Conference on Harmonization (ICH) guideline Q6A provides guidance (decision tree #3) on when a particle size specification should be considered [5]. In summary, a particle size specification is required if the particle size of the drug substance is critical to drug product performance (i.e., dissolution, solubility, bioavailability, content uniformity, stability, or product appearance) or drug product manufacturability (i.e. processability). However, in many New Drug Applications (NDAs) and Abbreviated New Drug Applications (ANDAs), control of particle sizes is considered for drug product performance only and the impact of particle sizes on drug product manufacturability (e.g., flowability, blend uniformity, and compactibility, etc.) frequently is not considered. For example, a particle size specification is established only for a poorly soluble drug substance due to the concerns of bioavailability or low dose drugs due to the concerns of content uniformity. If the drug product is not a low dose drug and the drug substance is highly soluble, the particle size specification may not be established or be established with little or no scientific justification.
For solid oral dosage forms, the impact of particle sizes of pharmaceutical powders on drug product manufacturability is of significant importance due to the fact that most of the unit operations for manufacturing of solid oral dosage forms are related to manipulation of the particle sizes of pharmaceutical powders. For instance, granulation and coating are related to particle size enlargement, while milling and grinding are related to particle size reduction. Screening and sieving are related to separation of particles with different sizes. Mixing and blending are related to combination of particles of different components where size differences among these components have a large impact on blend homogeneity. Therefore, understanding the impact of particle sizes of pharmaceutical powders on manufacturing processes is critical for each drug application. For raw or in-process materials, if the effects of their particle sizes on manufacturing processes (e.g., mixing, granulation, milling, blending, coating, etc.) are significant, control of PSDs of these pharmaceutical powders is necessary to ensure manufacturing consistency. Hence, the particle size specification is required for each of these materials, which may include not only the drug substance but also excipients as well as in-process materials. In the following, several manufacturing processes will be discussed to demonstrate when the particle size specification needs to be established for manufacturing of solid oral dosage forms.
Tablet Manufacture by Direct Compression
Direct compression is a process by which the tablets are compressed directly from powder blends of the active pharmaceutical ingredient (API) and suitable excipients (including fillers, disintegrants, and lubricants, etc.), which will flow uniformly into a die cavity and form into a firm compact [6]. For these different components (e.g., API, fillers, disintegrants, lubricants, etc.), if the differences in the particle size, shape, or density are significant, the powder blend (i.e., the mixture) may have a tendency to segregate, which will result in failure of blend uniformity. Segregation is particularly likely to occur in mixtures in which the components differ markedly in size, with differences in shape and density as secondary factors [7]. In addition, since the individual API and excipient particles are still present in the mixture, each of the components can behave as individual particles. Even though the final powder blend meets the acceptance criteria of blend uniformity, API may segregate or agglomerate after mixing and prior to compression due to the differences of particle size, shape, or density among different components in the mixture. Therefore, particle size specifications are required for both API and key excipients in order to ensure manufacturing consistency. In addition, particle shape information may be required if particle shapes have significant influence on mixing and flow properties of final powder blends. The particle shape effects on mixing and tabletting processes have been reported in the literature [8-9]
Tablet Manufacture by Granulation.
In addition to direct compression, tablets can be manufactured by either wet or dry granulation processes. Granulation is essentially one of the size enlargement processes, which can improve both flow and compression characteristics [6]. In a dry granulation process, the powder particles of API and diluents are aggregated under high pressure (either by slugging to form a slug or by roller compaction to form a ribbon) and then broken down to granules by milling and sieving before compression. In a wet granulation process, a liquid is added to the powder particles of API and diluents to produce agglomerates and then broken down to granules by drying, milling, and sieving before compression. In either case, since the properties of the individual API and diluent particles are masked (at least to a certain extent) by granulation, the quality of tablets is directly influenced by granules (mixtures of API and diluents) instead of the individual API and diluent particles. Therefore, controls of the size distribution and the flow property of granules are potentially critical for final blending and compression processes.
As pointed out in the FDA Guide to Inspection of Oral Solid Dosage Forms: Pre/Post Approval Issues for Development and Validation (1994), for granulation, a major physical parameter used to demonstrate equivalence between batches is the particle size profile of granules. This is particularly important for the comparison of the biobatch with production batches and also, when processes are modified or changed. The particle size profile will provide useful information for demonstrating comparability. The size and even the type of granule can affect the pore size in a tablet and have an effect on dissolution. For example, a dissolution failure for a coated tablet was attributed to a change in the milling screen size, yielding a granulation with larger granules and resulting in slower dissolution [10]. Hence, the particle size specification for the granules is recommended for a granulation process. Since there is a pre-mixing process during the wet or dry granulation, control of PSDs of API and key excipients will be required if particle size effects of these components are significant on homogeneity of powder blend as well as the final tablets.
Extended-release Capsule Manufacture by Multi-layer Coating.

Figure 1 - Variations of pellet size distributions during multi-layer coating processes
Table 1 - Changes of pellet size distributions during multi-layer coating processes.
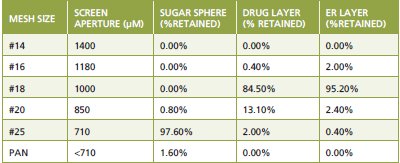
Many extended-release (ER) drug products are comprised of capsules filled with small, coated sugar pellets. Manufacture of these pellets involves drug layer coating and ER layer coating processes, which are essentially size enlargement processes. An example of growth of pellet sizes during multi-layer coating processes is shown as Figure 1. Monitoring and control of variations of pellet sizes at different coating stages may provide an effective way for control of the drug product quality. Heinicke et. al. studied the correlation between the pellet size and the coating thickness [11] and demonstrated that the polymer coating thickness can be measured by dynamic imaging analysis (DIA), which provides a potential tool as a surrogate dissolution test for a coated multiparticulate product [12]. In addition to imaging analysis, even a simple sieving analysis can be used to assess the variations of pellet sizes at different stages quantitatively. Table 1 presents an example to demonstrate how the pellet size distribution is changed at different manufacturing stages. These pellet size profiles measured at different stages provide useful information for comparison of batch to batch consistency. Furthermore, since a broad pellet size distribution may result in pellet segregation and affect content uniformity, control of pellet size distribution is critical not only for coating but also for blending processes. It is recommended to establish a particle size specification with appropriate acceptance criteria for initial sugar pellets, drug layered pellets, ER coated pellets, and final blended pellets for the multi-layer coating process. With regard to the drug substance, a particle size specification is not generally required if the drug substance is totally dissolved in the coating solution. However, if the drug substance is suspended in the coating solution, a particle size specification should be established as well.
In addition to control of pellet size distributions, coating uniformity for both the drug layer and the ER layer is critical for the drug product quality. Due to the small size of pellets, measurement of weight gain is not meaningful for control of coating uniformity. Instead, for each coating stage, coating uniformity should be assessed by assay and content uniformity of API for the unit-dose samples collected by an appropriate sampling plan at multiple locations. When the mean assay value of API is significantly different from the expected quantity, or there are large between-location variances of assay values, the coating process is probably not under control. Similarly, the in-process dissolution testing of samples collected by an appropriate sampling plan at multiple locations within the final blender or container before encapsulation is a useful tool for assessment of coating uniformity of the ER layer.
How to Establish an Appropriate Particle Size Specification?
Once it is determined that a particle size specification of pharmaceutical powder is needed, the next question is how to establish an appropriate particle size specification. In many NDAs and ANDAs, it is found that the particle size specifications submitted are inadequate to control the PSDs of pharmaceutical powders. Therefore, it is important to understand what information should be included in the particle size specification to meet the regulatory requirement. Per ICH guideline Q6A, a specification is defined as a list of tests, references to analytical procedures, and appropriate acceptance criteria, which are numerical limits, ranges, or other criteria for the tests described [5]. In the following, the establishment of an appropriate particle size specification, including analytical procedures, method validation, and acceptance criteria, will be discussed. Since sieving and laser diffraction are the most common methods used in drug applications for solid oral dosage forms, the discussion will mainly focus on these two particle sizing methods.
Analytical Procedure
With regard to the analytical procedure, United States Pharmacopeia (USP) general chapters <786> and <429> have provided useful information and requirements for the sieving and the laser diffraction methods respectively. However, the USP requirements are not fully addressed or followed in many submissions.
In general, particle size analysis by the sieving or laser diffraction method includes the following steps: (1) sampling of bulk powders, (2) sub-sampling of bulk samples for specimen, (3) specimen preparation or dispersion, (4) instrument set-up and verification, (5) size measurements, (6) data analysis and interpretation, and (7) report of size results [13]. Therefore, a complete analytical procedure should include all these information. During these stages, how to get a small amount of specimen with a representative PSD from the bulk powders is critical. Allen has demonstrated that selecting an appropriate sampling device will greatly improve reproducibility of the particle size measurements [14]. In addition, how to select an appropriate sample dispersion method is critical for sizing of small or cohesive particles which have a tendency to form agglomerates. The goal of sample dispersion is to eliminate as much particle agglomeration as possible from the sample analyzed and at the same time to avoid particle attrition or milling due to use of excessive dispersion forces [3]. In general, most errors in size measurements arise through poor sampling or poor sample dispersion but not through instrument inadequacies. Therefore, for a particle size specification, it is recommended to describe in detail the steps of sampling and sample dispersion strategies in the analytical procedure section.
For the laser diffraction method, data analysis and interpretation is also important as inappropriate choice of the optical model (i.e., Mie or Fraunhofer theory) or of the values of the refractive index may result in significant bias of the resulting particle size distribution [15]. Therefore, it is essential that the optical model and the refractive index values used are reported in order to obtain reproducible results. Since several significant differences exist, both in hardware and software, not only among instruments from different vendors but also among different types from one vendor, it is recommended to provide adequate information with regard to the laser diffraction instrument as well as software used into the analytical procedure section.
Method validation
Validation of an analytical procedure is the process by which it is established, by laboratory studies, that the performance characteristics of the procedure meet the requirements for the intended analytical applications [16]. Due to unique features of particle sizing methodologies, validation of particle sizing methods is not the same as validation of other analytical methods that are described in the ICH guidelines Q2A and Q2B and USP general chapter <1225> [17-18]. Generally speaking, validation of the particle sizing method usually involves evaluation of precision (repeatability and intermediate precision) and robustness. Other ICH defined validation parameters, such as specificity, linearity, range, accuracy, detection limit, and quantitation limit are not normally required for validation of the particle sizing method.
For validation of precision, repeatability refers to the use of the analytical procedure within a laboratory over a short period of time using the same analyst with the same equipment. Intermediate precision (also known as ruggedness) is a measure of variation within a laboratory, as on different days, or with different analysts or equipment within the same laboratory [16]. If the particle sizing method will be transferred to other laboratories, evaluation of reproducibility which refers to the use of the analytical procedure in different laboratories is required.
Robustness of an analytical procedure is a measure of its capacity to remain unaffected by small, but deliberate variations in method parameters and provides an indication of its reliability during normal usage [16]. Evaluation of robustness should be considered during the development of the particle sizing method. Once the optimal method parameters are determined, robustness can then be evaluated by deliberate small variations of these parameters. For the sieving method, the following parameters are recommended for validation of robustness: (1) sample mass, (2) tap rate, (3) rotation rate, and (4) agitation time. For the laser diffraction method, the following parameters are recommended for validation of robustness: (1) measurement stability, (2) refractive index, (3) dispersion pressure (for the dry measurement), and (4) sample concentration, sonication, and stir rate (for the wet measurement). It should show the reliability of an analysis with respect to deliberate variations in method parameters. A study should also be included in the method validation report to demonstrate that the sampling and sample dispersion strategies described in the analytical procedure are reliable and reproducible. In addition, it is recommended to use the imaging analysis as a complimentary tool to assess the particle shape, size range, and the suitability of the selected sieving or laser diffraction methods.
Acceptance Criteria
Acceptance criteria are numerical limits, ranges, or other suitable measures for acceptance of the results of analytical procedures [5]. For a particle size specification, the acceptable size range should be linked to drug product performance or drug product manufacturability. In general, a narrow PSD of pharmaceutical powders is desired. For a broad PSD, it is important to control the whole size distribution rather than the mean size only. Two powder systems with the same mean size but different size distributions may have significantly different influences on drug product performance or drug product manufacturability.

Figure 2 - A cumulative particle size distribution derived from a laser diffraction measurement
For the laser diffraction method, limits of D10, D50, and D90 are often used as the acceptance criteria. These parameters refer to the cumulative size distribution which describes how many percentages of samples are below a certain size of particles. D10, D50, or D90 is defined as the size value corresponding to cumulative size distribution at 10%, 50%, or 90%, which represents the size of particles below which 10%, 50%, or 90% of the sample lies. For example, size distribution data obtained form a laser diffraction measurement are plotted as a cumulative size distribution shown in Figure 2. It shows that D50 is 200 μm, which represents 50% of particles are below 200 μm (i.e., the median diameter). Similarly D10 and D90 are 74 μm and 411 μm, indicating that 10% of particles are below 74 μm and 90% of particles are below 411 μm respectively. It should be noted that, for the individual size measurement, each of D10, D50, and D90 is not a range but a size value. These values are changed at different measurements or different samples.
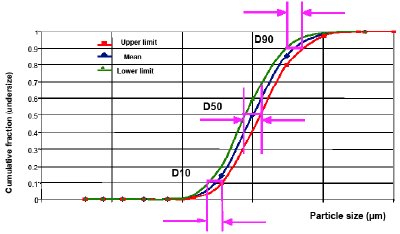
Figure 3 - Acceptance criteria of D10, D50, and D90 for the particle size distribution measured by the laser diffraction method. The blue line with diamond points represents the target or desired particle size distribution. The green line with triangle points represents the lower limit of the particle size distribution. The red line with square points represents the upper limit of the particle size distribution.
The PSD can be controlled by establishing both upper and lower limits for each of D10, D50, and D90. As shown in Figure 3, if the blue line with diamond points represents the target or desired PSD, the upper and the lower limits of D10, D50, and D90 provide a control strategy to ensure that variations of particle size distributions of samples between the green and red lines have not adversely affected the drug product performance or drug product manufacturability. Setting the values of the upper and lower limits will largely depend on how significant the effect of the PSD is, which is typically based upon the prior knowledge or design of experiment (DoE) studies. How to set these limits needs to be justified with supporting data, which should also be included in the drug application. In many NDAs and ANDAs, only the upper or the lower limit is established for D10, D50 or D90. This one-side limit is not acceptable without scientific justification since it does not provide adequate control for the PSD of powders.

Figure 4 - Acceptance criteria for the particle size distribution measured by the sieving method. The blue line with diamond points represents the target or desired particle size distribution. The green line with triangle points represents the upper limit of the particle size distribution. The red line with square points represents the lower limit of the particle size distribution.
For the sieving method, it is recommended to measure the whole particle size distribution of the sample rather than the proportion of particles passing or retained on one or two sieves. Once the cumulative size distribution is obtained from sieving measurements, the acceptance criteria can be established for D10, D50, and D90 by a similar approach used in the laser diffraction method. More often, the upper and lower limits of cumulative fractions for three sieve numbers (or particle sizes) are selected as the acceptance criteria as shown in the Figure 4. In this case, rather than control of size variations, the variations of cumulative fractions at three fixed sieves are controlled to ensure control of the PSD. Therefore, it is recommended to select three sieves of which the target cumulative factions are around 10%, 50%, and 90% in order to well-represent the whole particle size distribution.
Conclusions
This paper provides a regulatory perspective on the particle size specification for solid oral dosage forms. Since manufacturing of solid oral dosage forms is essentially related to manipulation of particle sizes of pharmaceutical powders, the impact of particle sizes of pharmaceutical powders is of significant importance. In addition for the drug substance, establishing appropriate particle size specifications for excipients as well as in-process materials may be necessary in order to ensure manufacturing consistency as well as the drug product quality.
With regard to the particle size specification submitted in a drug application, it is recommended to provide complete information in the analytical procedure including various aspects of sampling, sample preparation or dispersion, instrumental parameter setting, data analysis and interpretation, etc. A validation report for this particle sizing method should be provided as well including validation of repeatability, intermediate precision (ruggedness), robustness, etc. In addition, it is recommended to provide a study in the method validation report to demonstrate that the sampling and sample dispersion strategies described in the analytical procedure are reliable and reproducible. For the acceptance criteria, it is recommended to establish both the upper and the lower limits for better control of a broad particle size distribution. Justification with supporting data regarding how to set these limits should be provided in the submission. APR
Acknowledgement
The authors would like to thank Dr. Lawrence Yu and Gary Buehler for their valuable suggestions and comments.
References
- M.E. Fayed and L. Otten, Handbook of Powder Science & Technology, 2nd, Chapman & Hall, 1997.
- A. J. Hlinak, K. Kuriyan, K. R. Morris, G. W. Reklaitis, and P. K. Basu. Understanding critical material properties for solid dosage form design. Journal of Pharmaceutical Innovation, vol.1, 2006, pp.12–17.
- B.Y. Shekunov, P. Chattopadhyay, H.H.Y. Tong, and A.H.L. Chow, Particle Size analysis in Pharmaceutics: Principles, Methods and Applications, Pharmaceutical Research Vol. 24, No.2, 2006, pp.203-227.
- S.M. Snorek et al., PQRI Recommendation on Particle Size Analysis of Drug Substances Used in Oral Dosage Forms, Journal of Pharmaceutical Science, Vol. 96, No.6, 2007, pp.1451-1467.
- ICH Guideline Q6A, Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances, 2000.
- H.A.Lieberman, L. Lachman, and J.B. Schwartz, Pharmaceutical Dosage Forms: Tablets, 2nd, Marcel Dekker, 1989
- N.A. Armstrong, Tablet Manufacture, Encyclopedia of Pharmaceutical Technology, Marcel Dekker, 2002
- V. Swaminathan and D.O. Kildsig, Polydisperse powder mixtures: effect of particle size and shape on mixture stability. Drug. Dev. Ind. Pharm., vol. 28, 2002, pp.41-48.
- C. Sun and D.J. W. Grant, Influence of crystal shape on the tableting performance of i-iysine monohydrochloride dehydrate, Journal of Pharmaceutical Science, vol.90, 2001, pp.569-579.
- U.S. Food and Drug Administration, Guide to Inspection of Oral Solid Dosage Forms: Pre/Post Approval Issues for Development and Validation, 1994.
- G. Heinicke, F. Matthews, and J.B. Schwartz, The Effects of Substrate Size, Surface Area, and Density on Coat Thickness of Multi-Particulate Dosage Forms, Pharmaceutical Development and Technology vol.10. 2005, pp.85-96.
- G. Heinicke and J.B. Schwartz, Assessment of Dynamic Image Analysis as a Surrogate Dissolution Test for a Coated Multiparticulate Product, Pharmaceutical Development and Technology vol.11. 2006, pp.403-408.
- A. Jillavenkatesa, S.J. Dapkunas, and L.H. Lum, Particle Size Characterization, National Institute of Standards and Technology, Special Publication 960-1, 2001.
- T. Allen, Particle Size Measurement, 4th, Chapman and Hall, London, 1993.
- ISO 13320-1, Particle Size analysis-Laser Diffraction methods-Part 1: General Principles, International Organization for Standardization, 1999.
- The United States Pharmacopeia 30-The National Formulary 25, <1225>, Validation of Compendial Procedure, 2008.
- R. Bell, A. Dennis, B. Hendriksen, N. Norht, and J. Sherwood, Position Paper on Particle Sizing: Sample preparation, method validation, and data presentation, Pharmaceutical Technology Europe, November 1999, pp.36-42.
- S.A. Lerke and S.A. Adams, Development and Validation of a Particle Size Distribution Method for Analysis of Drug substance, American Pharmaceutical Review, Fall 2002.