Abstract
An analysis was conducted of 642 microbiologically-related recalls over the years 2004-2011. This analysis was conducted using publically available enforcement reports as presented on the US FDA website. The microbiologically-related recall activity shows a decided increase in recent years. Most of the reported recalls involved sterile products, and of these medical devices accounted for the majority. The reasons given for sterile product recalls were varied, but the majority cited “Lack of Sterility Assurance” with sterile packaging clearly identified as the main culprit.
There was significant information in the recall data for non-sterile products as well. The majority of the recalls came from OTC and personal care products, with “Objectionable Organisms” as the most prevalent reason for recall by a wide margin. These recalls are further analyzed to provide indication of the FDA policy on what is an objectionable organism, along with a review of current regulatory guidance. Finally, recommendations are presented in determining an “Absence of Objectionable Organism” policy for a manufacturer.
Introduction
In a previous report, one of the authors presented information on recalls in the US market that had a microbial basis [1]. A review of enforcement activity is important, as in addition to the Agency’s written policy on GMP there is a great deal of “corporate culture” to CGMP that is not written in official guidance documents but is nonetheless strictly enforced. By studying enforcement patterns we can deduce policy.
This report is an update on this topic, covering the years 2004 - 2011, with particular attention paid to non-sterile recalls. There will be some discussion of the sometimes confusing concept of “objectionable organisms” and what the analysis of product recalls can teach on this subject.
A note on the methodology used in this study is important. The source information is listed on the FDA web site, listing recalls by “Enforcement Reports.” These enforcement reports, while a valuable source of information, do have several limitations. First of all, the recalls are listed (on the Enforcement Report) by Recall Identification Number. This recall may include one batch, or several hundred batches. A single recall may also cover many different, but related products (and multiple batches of each product). The reader is urged to avoid the mistake of confusing the number of recalls with the eventual impact of that number on a particular company or on the industry. Secondly, there seems to be a significant and variable period of time between the actual event (the recall) and the appearance of the recall in the enforcement report. Therefore the dates of the enforcement reports should be viewed only as identification of the report, not necessarily as dates of recalls. For the purposes of this article we will, however, refer to the date of the enforcement report and the date of the recall interchangeable for simplicity’s sake.
The Enforcement Report lists several different categories of recalls. This review does not consider the differences among class 1, class 2 and class 3 recalls (refer to 21 CFR 7.41). The Enforcement Report format also catalogs the recalls by product type. With some slight variability over the years, the main product categories used in the Enforcement Reports include:
- Food (including personal care products)
- Drugs (OTC and prescription)
- Biologics
- Devices
- Veterinary
This review focuses on personal care products, drugs (pharmaceutical and OTC), and medical devices. The category of Biologics recalls was deliberately omitted despite its obvious interest to the readership because of the extraordinarily large number of recalls in virtually all weekly Enforcement Reports dealing with irregularities in the blood supply. Inclusion of this data would skew the results for the entire review.
Overview
The years 2004-2011 saw 642 recalls that involved a microbiological component, with a clear increase evident in 2010 and 2011 (Figure 1). This increase in microbiologically-related recalls reflects an increase in overall enforcement activity by FDA. In fact, 2009 might have also shown an increase in overall recalls however but the Agency was consumed by enforcement activities involving peanuts and pistachios. The evident dip in enforcement activity in 2009 for microbiologically-related issued might be due to a limitation of resources within the Agency.
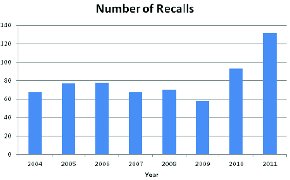
Figure 1 - Microbiologically-related Recalls by Year. Total number of microbiologically-related recalls as described in FDA weekly enforcement reports for the product categories included

Figure 2 - Microbiologically-related Recalls by Product Category. The authors determined the type of fi nished product involved in the microbiologically-related recall. Much of this information was supplied in the Enforcement Report (Food/Cosmetics, Drugs, Medical Device) and the authors used this information to assign these categories
As we look deeper into the microbiologically-related recalls we can see some distinct preferences in terms of types of products involved in these situations (Figure 2). Most of these recalls involve Medical Devices, followed by Pharma and OTC products. Dietary Supplements and Probiotics are the least, but this may be an artifact of the recent implementation of 21 CFR111, and the fact that with all the enforcement opportunities offered by the dietary supplements industry microbiological concerns are relatively low on the list.
Sterile Product Recalls
If we look at the data from a different perspective we can evaluate FDA concerns for Sterile vs Non-sterile products. Over ¾ of the recalls during the years 2004-2011 involved sterile products (Figure 3). Of these sterile product recalls, approximately 80% were due to “Lack of Sterility Assurance” (Figure 4) with remaining due to “microbial contamination”, a failed finish product test (BET or antimicrobial effi cacy), or was an issue with a diagnostic test (usually involving media or microbial ID kits) (Figure 5). The finding “Lack of Sterility Assurance” is frequently discussed at conferences and in publications, it is useful to look at what this really means from the perspective of enforcement activities.
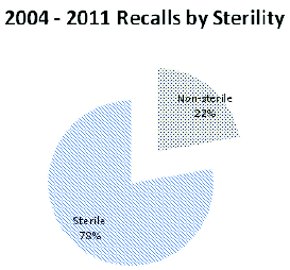
Figure 3 - Microbiologically-related Recalls by Product Sterility. Distribution of the included recalls based on the required state of sterility for the fi nished product
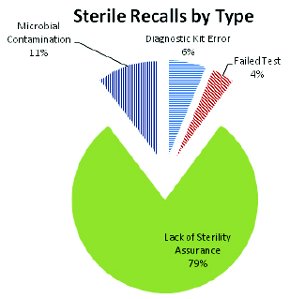
Figure 4 - Microbiologically-related Recalls of Sterile Products by Recall Category. One of fi ve recall categories were assigned to the product recalls for sterile products. Recalls for “microbial contamination” may or may not have a contaminating microorganism identifi ed; “Failed test” is either a failed sterility test, a failed antimicrobial preservative eff ectiveness test or bacterial endotoxin test (BET) associated with the recall; “Diagnostic kit error” is a broad category which includes media issues, biological indicator issues and microbial identifi cation kit issues (as well as others). Finally a recall would be placed in the “Lack of sterility assurance” if this is the cited reason on the enforcement report, or if the cited reason mentions product sterility but the recall (of the sterile product) does not fi t into any other category
Looking at the underlying causes of “Lack of Sterility Assurance” we can see that most of them are the result of packaging concerns (incomplete or weak seals, pinpricks in the sterile barrier, transport issues, etc). Of the remainder, almost all are either undetermined GMP issues or frank manufacturing errors (incomplete sterilization, non-sterile components added to sterile products, etc). Relatively few of these “Lack of Sterility Assurance” recalls actually showed contamination. It is more common to issue the recall on the basis of “contaminated product” (see Figure 4) rather than cite “Lack of...” for these situations. From this, it seems apparent that “Lack of Sterility Assurance” means either that there is a potential problem with the product or packages, or that the manufacturer cannot document that the product was manufactured and sterilized in a state of control. If the product is obviously contaminated, that is the cited reason for the recall (in the vast majority of cases).

Figure 5 - Underlying reason for “Lack of Sterility Assurance” citation in recalls. Those sterile product recalls were examined to determine the basis for a recall reason involving “Lack of Sterility Assurance.” Most were packing issues (pinpricks, defective seals, damage to the sterile barrier during transport, etc)

Figure 6 - Non-Sterile Recalls by Product Type. Non-sterile product recalls were most commonly either OTC or personal care products
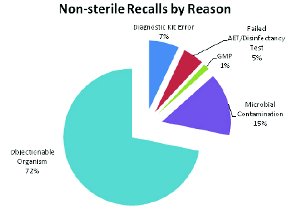
Figure 7 - Non-sterile Product Recalls by Reason
Non-sterile Product Recalls
An area of particular interest to the authors is the regulation of non-sterile medications. Looking at the enforcement reports for the non-sterile products, approximately ¾ of the recalls are due to either OTC products or personal care products during this period (Figure 6). The reasons for the non-sterile recalls are presented in Figure 7.
Figure 7 draws our attention to the topic of Objectionable Organisms. The issues facing non-sterile manufacturers are peculiar in that the finished product is intended to be contaminated (non-sterile). The challenge is to manufacture a non-sterile finished dosage form that does not have too high a level of contamination, and is not contaminated with the wrong type of organisms (objectionable ones).
The Code of Federal Regulations provides some guidance:
- 21 CFR 211.84(d)(6) “Each lot of a component, drug product container, or closure with potential for microbiological contamination that is objectionable in view of its intended use shall be subjected to microbiological tests before use.”
- 21 CFR 211.113(a) “Appropriate written procedures, designed to prevent objectionable microorganisms in drug products not required to be sterile, shall be established and followed.”
- 21 CFR 211.165(b) “There shall be appropriate laboratory testing, as necessary, of each batch of drug product required to be free of objectionable microorganisms.”
However, it must be noted that this does nothing to help determine what an objectionable organism might be.
Further guidance is provided by USP in the harmonized informational chapter <1111> [2]. It must be noted that the compendial Microbial Limits Tests are not intended to serve as a test for objectionable organisms [3] nor were they ever intended to do so [4,5].
Bringing this academic discussion into the real world, FDA has a solid track record of enforcing the requirement for absence of objectionable organisms in raw materials and finial, non-sterile products. In the previous review of recalls from 1998 through 2006 [1] 134 recalls listed on the FDA website for this time period were identified. Of these, only 14 were due to organisms listed in the Microbial Limits Tests - the others were “objectionable” but not “specified.” In this review, 144 recalls were identified for non-sterile products, only 8 of these recalls cite organisms listed in USP <62>.
However, returning to USP chapter <1111> we read:
“In addition to the microorganisms listed in Table I, the significance of other microorganisms recovered should be evaluated in terms of the following:
- The use of the product: hazard varies according to the route of administration (eye, nose, respiratory tract).
- The nature of the product: Does the product support growth?
- Does it have adequate antimicrobial preservation?
- The method of application.
- The intended recipient: risk may differ for neonates, infants, the debilitated.
- Use of immunosuppressive agents, corticosteroids.
- The presence of disease, wounds, organ damage.
Where warranted, a risk-based assessment of the relevant factors is conducted by personnel with specialized training in microbiology and in the interpretation of microbiological data. For raw materials, the assessment takes account of the processing to which the product is subjected, the current technology of testing, and the availability of materials of the desired quality.”
This compendial guidance provides the basis for an evaluation of potentially objectionable organisms by a competent, trained, professional microbiologist. It should also be noted that a risk assessment approach is encouraged. The first consideration should be total numbers of microorganisms present. An unfortunate fashion has arisen that argues immunocompromised patients are at increased risk for oral drugs, such that extremely tight total aerobic counts (10 CFU/G for tablets) must be established for the specification [6]. This argument completely ignores the fact that these same patients may each a pound of food a day (105-107 cfu/g, or 5x107-5x109 cfu/lb) without harm. It is important to have total bioburden limits set, but they must be defensible. The true concern with high levels of bioburden would be that they may well indicate a manufacturing process out of control, or that a spoilage organism is proliferating in your product. If the numbers of organisms in the product are not large, the question remains to determine if those organisms present are “objectionable”.
The two methods currently in use to determine objectionable organisms both use some form of risk analysis. The first is to determine all organisms (or families) that might cause a problem (using science, regulatory policy or both). The basic goal here is to form a list of “objectionables” – if the organism is not on the list, it is defined as acceptable. The problem with this approach is that if a new organism arises that was not initially considered, or that the inspector feels is objectionable that your list failed to include, there may be several batches of product placed at risk.
The second approach to determination of the absence of objectionable organisms is to analyze all microbial isolates found and determine if they fit a criteria for “objectionable.” The difficulty with this method is that it is extremely labor intensive. Perhaps a blending of the two would be most productive, with organisms categorized as either “objectionable” or “benign” based on the product presentation and target population. The placement of the organism into either category would require research, but this research would then be retained for future use.
Table 1 provides a summary of recalls, with information on the product involved, Recall Category and Specific Reasons for the recall (both deduced by the authors), the date of the enforcement report and a reproduction of the reason cited by FDA in the enforcement report. Based on the review of recalls, both numbers of organisms and the objectionable nature of some organisms have been responsible for FDA participation in “voluntary” (voluntary in the sense that the company accedes to the FDA recommendation) as well as enforced recalls. Of the 142 non-sterile recalls during the period 2004-2011 listed in Table 1, 103 were for objectionables and 22 for “microbial contamination” (Figure 7). As might be expected, the breakdown of the organism types is as follows:
- Gram Negative Bacilli – cited in 77 recalls
- Yeast/Mold – cited in 23 recalls
- Gram Positive Cocci – cited in 3 recalls
Table 1 - Non-sterile Recall Summary 2004-2011

Table 1 - Non-sterile Recall Summary 2004-2011 (cont.)

Table 1 - Non-sterile Recall Summary 2004-2011 (cont.)
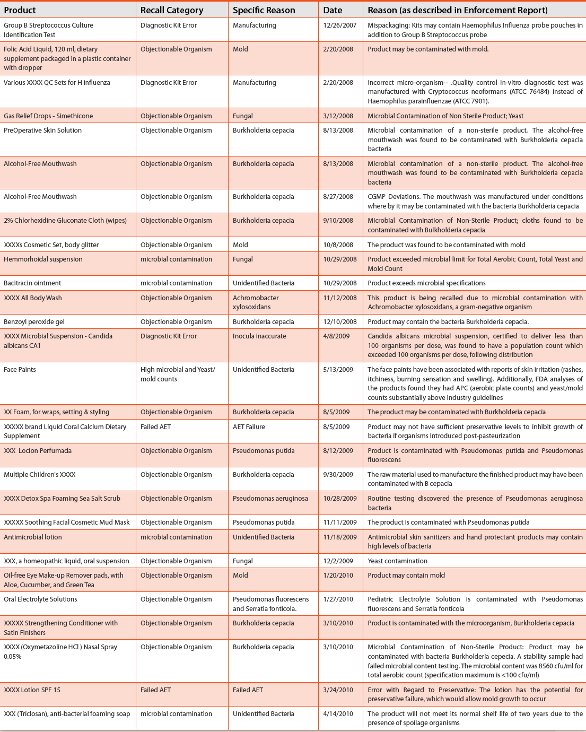
Table 1 - Non-sterile Recall Summary 2004-2011 (cont.)
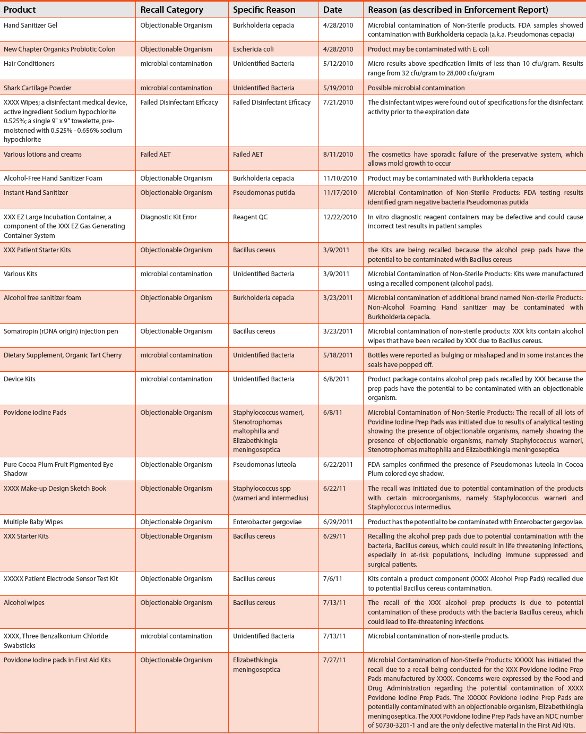
Table 1 - Non-sterile Recall Summary 2004-2011 (cont.)

Table 1 - Non-sterile Recall Summary 2004-2011 (cont.)

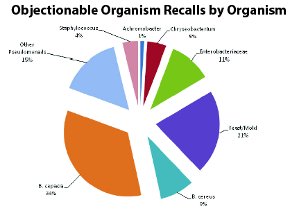
Figure 8 - Objectionable Organism Recalls by Organism for Non-sterile Products
Figure 8 provides details on the types of organisms cited in FDA enforcement reports for this period. From this analysis it is clear that the pseudomonads are the most frequently cited of the “objectionables” in recalls of this type. Table 2 provides detail on the cited identity of microorganisms in these recalls.
One of the immediate impressions from Table 2 is the prevalence of recalls that specifically cite Burkholderiacepacia as the objectionable organism in non-sterile products. In fact, B. cepacia alone is cited in 34% of the non-sterile recalls from the years 2004-2011. This extends a trend reported for the years 1998-2006 where B. cepacia was the cited cause for non-sterile recalls in 22% of the cases [1]. The recalls involving B. cepacia in the 2004-2011 timeframe have ranged from mouthwashes (alcohol- free mouthwash packaged both independently and in hospital hygiene kits), moist wipes, soaps and sanitizers, nasal products to hair dyes.
Table 2 - Identity of Objectionable Organisms Cited in Recalls for “Objectionable Organisms” 2004 – 2011

As discussed above, B. cepacia holds a special place in the corporate culture of FDA. This dates back to a tragedy in the early 1980s when an inhalant was marketed contaminated by Pseudomonas cepacia (Burkholderia cepacia). This product had passed USP tests (which are not capable of recognizing B. cepacia – see USP 1982 for more details). However, this event caused the death of several cystic fibrosis patients and lead to the realization that this organism had the capability to cause disease in a susceptible population and also to survive in preserved solutions [7,8]. This also led to the establishment of a requirement that aqueous-based inhalants must be sterile (21 CFR 200.51) [9].
Recently a rationale was published discussing the intense concern that the Agency continues to feel towards B. cepacia as an objectionable organism in a wide range of non-sterile products [10]. This article has raised additional questions from the field [11]. However, there is a real argument that can be made for concern over B. cepacia in non-sterile products that enter the nasal passage or the lungs, particularly in those populations susceptible to pneumonia. These patient populations at risk might include neonates, advanced elderly and cystic fibrosis patients (among others) and products marketed to those populations, or likely to be used on those populations, should not contain B. cepacia.
Given that the organism is a concern for severely compromised individuals if presented to the lungs, is this an argument for the elimination of B. cepacia from all non-sterile products? The Agency has stated from the podium that it is FDA policy to “regulate to the most susceptible population.” This policy strikes the authors as ill-advised. First of all, it is imprecise. The most susceptible population is that portion completely devoid of a functioning immune system (regulating to Bubble-boy is unworkable). Since this is clearly not what the Agency means, we are left to divine its intent based on clues. Secondly this policy is illogical, “regulating to the most susceptible population” might be as well expressed as “everyone in the family must eat strained carrots because the baby cannot eat steak.” In no other area of life or business would this philosophy be followed. It is far more reasonable and efficient to control the access to, and use of, products by the user, based on the risk posed to that user. There are those allergic to shellfish –yet the sale of shellfish continues. However, it is clearly the policy of the FDA that B. cepacia is a severe threat to the health of the nation and will be viewed as sufficient reason to encourage a “voluntary” recall if seen in a product (despite the paucity of scientific data supporting its hazardous nature in most situations). The large number of non-sterile product recalls during this period citing B. cepacia underscores the importance of this discussion.
Another troubling area for this analysis of non-sterile recalls is the large number (15%) that only cite “microbial contamination” as the cause for a non-sterile product recall. One must assume that many of these are due to extremely large numbers of microorganisms present, but this should properly have been cited for the record. Having these large numbers of microorganisms is objectionable in and of itself as it might either indicate slovenly manufacturing practices or the presence of a spoilage organism that is growing in the product. In either case, given the widespread availability of microbial identification systems [12] the failure to note (or to identify) the causative microorganism(s) is regrettable.
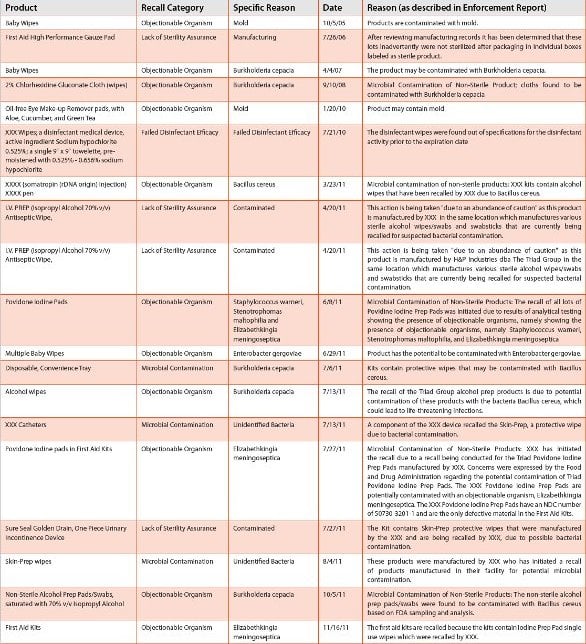
A final point of interest is the prevalence of Bacillus cereus in the listing. All of the 2011 recalls of this product category are linked to the recall of product (“sterile” and non-sterile alcohol prep pads) from a single manufacturer. There are two points here. The first is the enormous impact this recall had on many different products – both sterile and non-sterile (mainly kits). The second consideration is that FDA is clearly concerned with the current safety of moist wipes on the marketplace. A summary of recalls involving moist wipes is presented in Table 3. Perhaps the sterility requirements for this product category – moist wipes – needs to be reconsidered. Is there a place for non-sterile moist wipes in the marketplace? If so, should procedures that require sterile wipes (for example, preparation of an injection site) be clearly described to prevent the misuse of labeled non-sterile wipes?
Table 3 - “Wipes” Recall Summary 2004-2011
Where does this leave us in terms of generating an understanding of “objectionable organisms”? Much depends on your company’s scientific sophistical and tolerance of risk. Using the previous recall review with the data presented in this review and available regulatory documents one might put together a list of “objectionables”. This is probably the best course of action for those least tolerant of risk or desirous of engaging the Agency in scientific debate. For those who would have a rationale for this risk assessment, the authors urge a risk assessment strategy based on the rationale presented in USP chapter <1111>.
Conclusions
An analysis was conducted using publically available enforcement reports as presented on the US FDA website. The microbiologically-related recall activity shows a decided increase in recent years. Most of the reported recalls involved sterile products, and of these medical devices accounted for the majority. The reasons given for sterile product recalls were varied, but the majority cited “Lack of Sterility Assurance” with defects and weaknesses of sterile packaging clearly identified as the most common source of contamination potential.
There was significant information in the recall data for non-sterile products as well. The majority of the recalls came from OTC and personal care products, with “Objectionable Organisms” as the most prevalent reason for recall. Information on FDA policy in terms of “objectionable organisms” was apparent from the data.
This review demonstrates that analysis of enforcement reports, freely available from the FDA web site, provides insightful and actionable information on CGMP. A review of this kind also makes one extremely grateful for the diligence of the FDA in safeguarding the regulated industries.
References
FDA. 2011. Enforcement Report Home Page. http://www.fda.gov/Safety/Recalls/EnforcementReports/default.htm Accessed December, 2011.
- Jimenez, L. 2007. Microbial diversity in pharmaceutical product recalls and environments. PDA Journal of Pharmaceutical Science and Technology 61(5):383-399.
- USP. 2011.<1111>Microbiological Examination Of Nonsterile Products: Acceptance Criteria For Pharmaceutical Preparations And Substances For Pharmaceutical Use USP 34/NF 29 vol. 1. United State Pharmacopeial Convention pp. 630-631.
- Sutton, S. 2006. The Harmonization of the Microbial Limits Tests Pharm Technol. 30(12):66-73
- FDA. 1993. Guide to Inspections of Microbiological Pharmaceutical Quality Control Laboratories. http://www.fda.gov/ICECI/Inspections/InspectionGuides/ucm074914.htm.
- USP. 1982. Microbial Contamination of Sterile and Non-Sterile Articles, with Special Reference to Pseudomonas cepacia. Pharm Forum 8(4):2239.
- Manu-Tawiah, W et al. 2001. Setting Threshold Limits for the Significance of Objectionable Microorganisms In Oral Pharmaceutical Products. PDA J Pharm Sci Tech. 55(3):171-175
- Kuhn, RJ et al. 1982. Bacterial Contamination of Aerosol Solutions Used To Treat Cystic Fibrosis. Amer J Hosp Pharm 39:308-309.
- Decicco, B., Lee, E., et al. 1982. Factors Affecting Survival of Pseudomonas cepacia in Decongestant Nasal Sprays Containing Thimerosal as Preservative. J Pharm Sci. 71(11):1231 - 1234
- FDA. 2002. Guidance for Industry: Nasal Spray and Inhalation Solution, Suspension, and Spray Drug Products — Chemistry, Manufacturing, and Controls Documentation
- Torbeck, L., et al. 2011. Burkholderia cepacia: This Decision Is Overdue. PDA J Pharm Sci Tech 65(5):535-543
- Sutton, S. 2012 Commentary on “Burkholderia cepacia: This Decision Is Overdue”. PDA J Pharm Sci Tech In Press.
- Cundell, AM. 2006. Microbial Identification Strategies in the Pharmaceutical Industry PDA J Pharm Sci Tech 60(2):111-123.