Abstract
Polymorphs, solvates, inclusion complexes and co-crystals of active pharmaceutical ingredients (APIs) are four established classes of ‘supramolecularly derived’ therapeutic agents. Each distinct solid phase in any of these classes contains a given API in an intact molecular form and possesses a unique crystalline structure and hence unique physical properties that warrant careful consideration and monitoring during its isolation, processing, formulation, delivery and storage.
Hitherto unknown solid species in these classes are constantly being sought by pharmaceutical scientists because they present new opportunities for drug development. Physicochemical characterization of each new solid form of an API to establish its structure, thermodynamic stability and its relationship to known forms is an essential part of the preformulation phase. Thermal analysis plays a critical role in such characterization, but unequivocal interpretation of the derived experimental data relies on insights afforded by complementary techniques such as spectroscopy (FTIR, Raman, solid-state NMR) and X-ray diffraction. In this review, several recently reported studies highlighting the application of thermal analysis to the four classes of pharmaceutically relevant solids are presented; these studies also reflect the interdependence of characterization methods when applied to ‘supramolecularly derived’ species.
Introduction
The structural uniqueness of each crystalline form of a given API confers a unique set of physical properties on that phase. Consequently, selection of the ‘right’ solid form of an API is crucial for optimizing the various phases of drug development including its isolation, processing, formulation and storage. Judicious choice of the appropriate solid-state form of the API is tantamount to ‘tuning’ critical technological and thermodynamic properties, resulting in one or more significant improvements, such as facile tabletability, reduced hygroscopicity, increased thermal stability and improved aqueous solubility [1-2]. This background provides strong motivation for the discovery and development of new solid-state forms of APIs and their exploitation as alternatives to existing forms, practices that are being avidly pursued worldwide.
The focus here is on solid-state forms in which the molecule of the API is not compromised by covalent modification (as occurs instead in prodrug synthesis which involves making and breaking covalent bonds in the API molecule, or in salt formation, with transfer of a proton from one reacting species to the other). Diff erent polymorphic forms of the API are one-component systems of this type, while API solvates, cyclodextrin- API (CD-API) inclusion complexes, and co-crystals of APIs represent three categories of multi-component systems. In all cases, these solid- state forms are supramolecular assemblies in which neutral molecules are bound to one another via ‘soft’ interactions (e.g. hydrogen bonding, hydrophobic interactions, π-stacking), no covalent bonds being made or broken during their formation and subsequent crystallization. The four types of products referred to here may thus be considered as supramolecular derivatives of the API. This concept is illustrated for the drug acetaminophen as an example (Figure 1), the variously derived species indicated being authentic, realizable entities. Our own ongoing research program has been based on this supramolecular approach to generating multiple, novel forms of APIs.
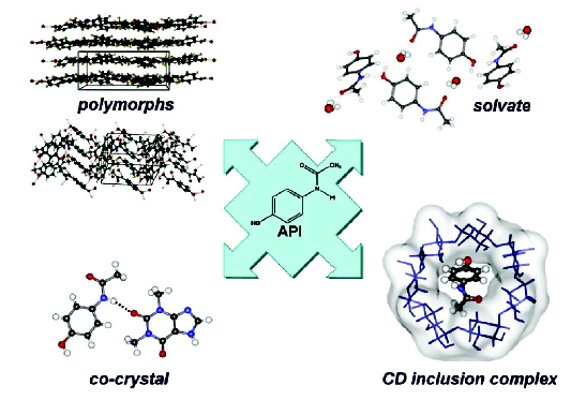
Figure 1 - Supramolecular derivatisation of an active pharmaceutical ingredient.
The very signifi cant diff erences in the packing arrangements of acetaminophen molecules evident in the two polymorphs illustrated in Figure 1 have genuine practical implications, the ‘graphitic’ (layered) form being easily compressed into tablets while the second polymorph requires addition of excipients for tableting [1]. Solvate formation, on the other hand, involves incorporation of solvent molecules by the API to stabilize the resultant crystal structure. In Figure 1, the included solvent, water, renders the resulting monohydrate phase of the API a pharmaceutically acceptable entity. Complex formation between the API and a biocompatible cyclodextrin [specifi cally β-CD, in the example in Figure 1, a cyclic oligosaccharide consisting of seven α-1,4-linked glucose molecules] likewise involves only non-covalent interactions between the host (CD) and the guest (API), the latter molecule occupying the hydrophobic micro-environment of the natural cavity of the CD molecule [3]. The fi nal species illustrated in Figure 1 is that of a co-crystal, a multi-component system generally comprising the API and one or more molecules of a co-former [a GRAS (‘generally regarded as safe’) and usually inactive partner] bound to one another by soft interactions [4]. In fact, the species shown, while technically qualifying as a co-crystal, is atypical in that the GRAS co-former in this case is the theophylline molecule, rather than an inactive species. It may therefore also be considered as a ‘combination drug’ candidate. Here, the two molecular components are linked by a predictable, well-defi ned N-H-- -O hydrogen bond, a typical feature of a ‘designed’ co-crystal.
Given the variety of solvents, CD derivatives and especially the large number of potential co-former molecules available, it is not diffi cult to imagine literally hundreds of supramolecular derivatives being generated from a single API, at least in principle. The number of potential solid forms derivable from the API is capable of yet further signifi cant increase since each solvate, co-crystal and CD inclusion complex could itself conceivably exist in various isolable polymorphic forms, while the range of co-crystals can be realistically extended through their conversion into solvates and into alternative co-crystalline variants by changing the stoichiometric ratio of API and co-former molecules. High-throughput screening is eff ective in exploring this rich potential diversity of solid form.
Each unique supramolecular construct has the potential to display novel properties and advantages for API development and each therefore requires detailed physicochemical characterization at the preformulation stage. Thermal analysis [commonly including hot stage microscopy (HSM), thermogravimetric analysis (TGA) and diff erential scanning calorimetry (DSC)] are essential for determining fundamental characterization data for such novel solid forms. These data include inter alia accurate melting points, solvent mass losses and desolvation temperature ranges for solvated species, temperatures and enthalpies of polymorphic transformations, polymorphic relationships (enantiotropy, monotropy) and kinetic data for thermal degradation to enable predictions of product shelf life. Some studies that highlight contemporary use of thermal analysis in such applications are described in subsequent sections.
Polymorphs and Solvates of APIs
There is a vast literature on the applications of thermal analysis to studies of polymorphs and solvates, including numerous reviews, some recent ones being referenced here [5-7]. Owing to space limitations, only a handful of individual reports that give some fl avor of recent studies in this area are cited explicitly below. While amorphous phases of APIs are not explicitly discussed in the context of this article, they are nevertheless highly relevant in pharmaceutical development, generally being signifi cantly more soluble than their crystalline counterparts [1-2], but having a tendency to crystallize spontaneously. Reference to the amorphous state does, however, appear in some of the studies cited here.
Discovery of a new polymorph of an API immediately raises the question as to its thermodynamic relationship to known polymorphs of the same material. A thorough investigation would typically involve preliminary examination by HSM to characterize crystal morphology and general behavior on heating. Recording the DSC trace would reveal the melting point and the corresponding enthalpy of fusion, as well as possible additional endotherms and exotherms arising from polymorphic transformations. Comparison of these features with those for known polymorphs would establish the various thermodynamic relationships (enantiotropic, monotropic) among all the available forms, as well as their stability ranking, based on their respective Gibbs free energy (G) values. Ideally, a quantitative or semi-quantitative energy-temperature diagram (G-T) incorporating all of this information would be constructed [1]. Such a diagram is invaluable, allowing predictions of all possible polymorphic interconversions during the course of heating or cooling a given polymorph. The thermodynamic stability ranking at any desired temperature, reflected in the respective Gibbs free energies of the various polymorphs, is of pivotal importance: the solid form with the lowest value of G at a given temperature and pressure is the most stable one and hence cannot spontaneously transform into any of the other known forms. In utilizing DSC in such investigations, it is usually necessary to confirm assignments of the appearance or disappearance of specific polymorphs by independent methods. Powder X-ray diffraction (PXRD), employed in static or dynamic (variable-temperature) modes, is the most powerful adjunct in this regard, enabling each distinct polymorphic phase to be ‘fingerprinted’. Such fingerprints can be directly correlated with the corresponding single crystal X-ray structures of the polymorphs, where these are available, or with crystal structures determined directly from high-resolution PXRD data [8].
High-throughput screening for polymorphs of an API by recrystallization from a range of solvents frequently yields one or more solvates. As noted earlier, additional features of their thermal analysis include the characterization of desolvation events by TGA and DSC techniques.
Comprehensive studies of polymorphic systems aimed at identifying the best candidate for formulation are fairly common, given their importance for pharmaceutical development. A recent representative paper reported that DSC and PXRD techniques were employed separately, and in combination in simultaneous dynamic DSC/PXRD mode, to elucidate stability relationships among ten distinct forms of an organic compound [9]. These forms comprised both unsolvated forms (polymorphs) as well as several hydrates (ranging from a monohydrate to a heptahydrate). A complex, but highly informative profile of the interconversions among these various solid forms was unravelled using DSC to evaluate the thermal parameters for transitions and dehydration processes, as well as their nature (reversible/irreversible), while PXRD provided unequivocal identification of the relevant forms involved. Taking all of the derived data into account, one of the anhydrous forms (Form VI) melting at 245°C was identified as the ideal species for further development.
In another representative case, polymorphic screening of the compound {4-(4-chloro-3-fluorophenyl)-2-[4-(methyloxy)phenyl]-1,3-thiazol-5-yl} acetic acid yielded two polymorphs [10] which were characterized by a variety of techniques including thermal analysis, PXRD, vibrational and solid-state NMR spectroscopy. Polymorphic Forms 1 and 2 were shown to be enantiotropically related, with, however, a relatively low thermodynamic transition temperature of 35 ± 3°C, the stable polymorph at room temperature being Form 1. In this instance, however, since the authors also established that milling of Form 1 resulted in its transformation to Form 2, the latter polymorph, though metastable at room temperature, was nevertheless the one selected as the candidate for further development. This decision necessitated close analytical monitoring of the processed material intended for drug loading to ensure minimum subsequent contamination from polymorphic Form 1. This example emphasizes that the thermodynamic stability of a particular solid phase at a given temperature cannot be viewed in isolation as a criterion for its safe development and that a judicious choice should be made on the basis of data gleaned from multiple techniques.
Establishing whether two polymorphs are monotropically or enantiotropically related relies on accurate measurement of their melting points and enthalpies of fusion [1], usually obtained by DSC. However, observation of the relevant transitions may be masked by an inappropriate choice of heating rate. Accelerated heating influences the kinetics of thermodynamic transitions and consequently can have profound effects on the appearance of the DSC trace, leading to separation of composite physical events. The advantage for studying metastable polymorphs, illustrated earlier with the drug carbamazepine [1], has been exploited more recently for a development compound by using DSC heating rates as high as ~500K/min. In this system [11], on heating the metastable form C at e.g. 10 K/min, melting of C and concurrent recrystallization to a second polymorph A were observed, together with a corresponding endotherm for the melting of A. However, at heating rates of 400 K/min and higher, the recrystallization process was suppressed, ensuring that only the melting endotherm for polymorph C appeared in the DSC trace, thus enabling its melting point and enthalpy of fusion to be determined accurately.
For technical reasons, modulated DSC (MDSC) has not been of major interest for investigating polymorphic transitions [1], but it has enjoyed increasing application in the estimation of the amorphous content of drug samples. A recent study involved the antihypertensive API carvedilol using both MDSC and PXRD to estimate amorphous content in bulk crystalline material [12]. For this system, the former technique was shown to be considerably more sensitive, especially at low levels of amorph. A comparative study of the application of Raman spectroscopy, dynamic mechanical analysis (DMA) and MDSC to the estimation of a minor amorphous phase in crystalline etoricoxib also appeared recently [13], limits of detection of the amorph being reported as 2, 2.5 and 5% respectively for these techniques.
TGA coupled with DSC is essential for distinguishing (anhydrous) polymorphs and solvates of APIs. Solvent content in the latter species may be stoichiometric or non-stoichiometric [1]. An interesting recent case of crystalline non-stoichiometric solvates containing the API nevirapine was encountered [14]. This API was found to form a series of isostructural solvates with primary alcohols CH3(CH2)nOH (n = 1-7) in which the molar amount (m) of alcohol in their nevirapine/alcohol molar ratios 1:m derived from accurate TGA measurements varied linearly from 0.54 to 0.32 with increasing alcohol chain-length, an unusual feature that defied explanation using only thermal analytical methods. A definitive explanation was eventually furnished by single crystal XRD and PXRD studies which revealed a common isostructural hydrogen-bonded nevirapine molecule framework in this series of solvate crystals. This isostructural framework provides a channel with constant topology and average periodicity 8.5 Å (the length of the crystal b-axis), which accommodates the respective solvent molecules. In the case of the 1-butanol solvate (n = 3), single crystal XRD showed that the solvent molecule (disordered over a centre of symmetry) spanned the b-axis length comfortably, giving rise to a commensurate crystal structure and a 1:0.50 (or 2:1) API:1-butanol stoichiometric ratio, while all the other alcohol molecules yielded incommensurate structures and compositions that are consequently characterized by non-integral stoichiometric coefficients.
Recent accounts of various aspects of the thermoanalytical behavior of drug solvates include those of APIs sulfamerazine [15], N4- acetylsulfamerazine [16], rifampin [17], and theophylline-7-acetic acid [18]. Variable-temperature PXRD to complement TGA and DSC analyses of polymorphs and solvates of APIs [19] continues to find wide application. The utility and importance of such techniques in phase and habit selection for a development compound have also been highlighted recently [5].
Cyclodextrin-API Inclusion Complexes
Owing primarily to the potential of CD inclusion technology to increase API solubility, complexes in this class are popular candidates for drug development and as such, their solid-state characterization is no less crucial than that for the polymorphs and solvates of APIs discussed earlier. The composition of the inclusion complex usually depends on whether the host compound is a native CD (α-, β-, or γ-CD) or a derivatised CD (e.g. the fully methylated equivalents). When aqueous media are employed for their synthesis, CD-API complexes of native CDs (containing exclusively free hydroxyl groups) are generally ternary species comprising the host, API (guest) and water molecules, the latter typically engaging in very extensive hydrogen bonding with one another and with CD hydroxyl groups to maintain crystal stability. Methylation of a native CD results in some or all of the –OH groups being converted to –OCH3 groups, reducing the hydrogen bonding capacity of the host and leading to inclusion complexes with reduced water content. Fully methylated CDs frequently include APIs to form crystalline inclusion complexes which are anhydrous. Furthermore, while inclusion complexes of native CDs typically decompose on heating, for some inclusion complexes of e.g. methylated CDs, a distinct melting point exists.
TGA analysis of CD inclusion complexes is frequently used for the estimation of crystal water content (typically ~10-20 water molecules per native CD host molecule and ~0-5 in the case of methylated CDs). TGA and DSC traces of these complexes usually indicate the multi- step nature of the dehydration process. In some instances, the API molecule may itself be released upon heating the inclusion complex, either concomitantly with water, or separately at a somewhat higher temperature, enabling also the CD:API molar ratio to be determined by TGA if this is not already known from other techniques (e.g. elemental analysis, NMR spectroscopy).
The versatility of CDs as hosts for APIs is refl ected in the large number of structurally diverse guest molecules that they are able to include within their cavities, either partially or completely [3]. Studies published during the last three years that refl ect the use of thermal analysis in solid-state characterization of CD complexes include those of the APIs fl uconazole [20], fosinopril [21], hydrochlorothiazide [22], lansoprazole [23], trimethoprim [24], meloxicam [25], spironolactone [26], nimodipine [27], cefdinir [28], valsartan [29], atenolol [30], thymoquinone [31] and fl uorofenidone [32]. A wide variety of CDs also features among these studies, including native host compounds, randomly methylated β-CD (RAMEB), and hydroxypropyl-β-CD (HPBCD). It should be mentioned that other macrocyclic molecules (e.g. calixarenes) have been employed to some extent in attempts to increase drug solubility. An account of the thermal analysis of complexes between water-soluble sulfonated calixarenes and the drugs nifedipine and niclosamide has appeared in this journal [33].
API Co-crystals
A distinguishing feature of the co-crystal approach to generating new pharmaceutically relevant solids is that their molecular design is based on rational principles of ‘crystal engineering’, entailing in the fi rst instance the identifi cation of appropriate functionalities on the API and co-former molecules that have the propensity to associate by predictable hydrogen bonding (Figure 2).
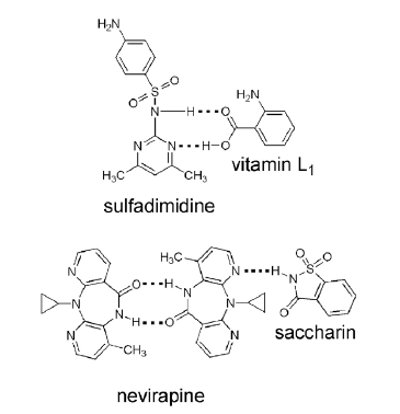
Figure 2 - Chemical structures of representative API co-crystals synthesized in the author’s laboratory [34-35].
According to a recent authoritative review of API co-crystals [36], progress in this fi eld has reached a stage where it is now possible to design such compounds, synthezise them by a variety of methods, characterize them and evaluate their properties; we are currently entering the ‘second phase’ in their development. The latter, according to the article cited, ‘involves the partial rationalization of observations made in the fi rst phase and the generation of empirical rules to predict properties of co-crystals’. As far as the application of thermal analysis is concerned, one of the properties of API co-crystals of primary technological interest is their thermal stability, determined by their lattice energy and refl ected in their melting points. The review cited above refers to a number ofrecent studies that indicate definite trends in co-crystal melting points whose systematic investigation could contribute to the future goal of more accurate prediction of this property. From several studies, it appears that the melting points of the co-crystals of a specific API show some tendency to follow the melting trend of the respective pure co-former compounds [4, 36]. In broad terms, if a higher melting form of a particular API is sought, it is likely to be attained by co-crystallizing the API with a high-melting co-former compound. The data supporting these generalizations have generally been accurately measured using the DSC technique. For example, in a study of ten co-crystals of a development compound with GRAS carboxylic acid co-formers, a good correlation was established between the DSC onset melting points of the co-crystals and those of the pure co-formers [37]. It remains to be seen whether more robust predictability will emerge from statistical treatment of more data of this type.
Summary
This brief account has focused on the application of (primarily) TGA and DSC thermal methods and supporting techniques to the characterization of new solid forms of APIs that are derived by a ‘supramolecular’ approach. This strategy of exploiting the various classes of the derivatives described above as serious candidates for API development has proven to be a viable one and it is likely to place ever-increasing pressure on thermoanalytical facilities for solid-state characterization, especially in view of the increasing tendency to employ high-throughput screening to generate candidate compounds. Some of the case studies cited indicate the importance of complementing the results of thermal analysis with data from supporting techniques.
Acknowledgements
The author is grateful to the University of Cape Town and the National Research Foundation (Pretoria) for research support.
References
- R. Hilfiker, “Polymorphism in the Pharmaceutical Industry“, Wiley-VCH 2006, Weinheim
- S. R. Byrn, R. R. Pfeiffer and J. G. Stowell, “Solid-state Chemistry of Drugs”, SSCI, Inc., 1999, West Lafayette, Indiana, USA.
- “Cyclodextrins and Their Complexes”, H. Dodziuk ed., Wiley-VCH 2006, Weinheim
- N. Schultheiss and A. Newman, Cryst. Growth Des., 2009, 9, 2950.
- T. Detoisien, M. Arnoux, P. Taulelle, D. Colson, J.P. Klein and S. Veesler, Int. J. Pharm., 2011, 403, 29.
- S. P. Stodghill, American Pharmaceutical Review, 2010, 13(2), 29, 31, 3-36.
- D. K. Murphy and S. Rabel, In: “Preformulation in Solid Dosage Form Development”, C. Adeyeye, and H. Brittain, eds.; Informa Healthcare USA, Inc., 2008, New York, USA.
- W. I. F. David and K. Shankland, Acta Crystallogr., Section A: Foundations of Crystallography, 2008, A64, 52.
- D. Albers, M. Galgoci, D. King, D. Miller, R. Newman, L. Peerey and E. Tai, Org. Process Res. Dev., 2007, 11, 846.
- L. M. Katrincic, Y. T. Sun, R. A. Carlton, A. M. Diederich, R.L. Mueller and F. G. Vogt, Int. J. Pharm., 2009, 366, 1.
- C. McGregor and E. Bines, Int. J. Pharm., 2008, 350, 48.
- D. M. Raut, D. M. Sakharkar, P. S. Bodke and D. T. Mahajan, Pharmacia Lettre, 2011, 3, 1.
- S. Clas, K. Lalonde, K. Khougaz, C. R. Dalton and R. Bilbeisi, J. Pharm. Sci., 2012, 101, 558.
- N. Stieger, W. Liebenberg, J. C. Wessels, H. Samsodien and M. R. Caira, Struct. Chem., 2010, 21, 771.
- S. Aitipamula, P. S. Chow and R. B. H. Tan, CrystEngComm, 2012, 14,, 691.
- S. Aitipamula, P. S. Chow and R. B. H. Tan, J. Mol. Struct., 2011, 1005, 134.
- M. M. de Villiers, M. R. Caira, J. Li, S. J. Strydom, S. A. Bourne and W. Liebenberg, Mol. Pharmaceutics, 2011, 8, 877.
- A. Foppoli, L. Zema, A. Maroni, M. E. Sangalli, M. R. Caira and A. Gazzaniga, J. Therm. Anal. Calorim., 2010, 99, 649
- S. X. Yin, R. P. Scaringe, M. F. Malley and J. Z. Gougoutas, American Pharmaceutical Review, 2005, 8, 56-58, 60, 62, 67.
- G. Yurtdas, M. Demirel and L. Genc, J. Incl. Phenom. Macrocycl. Chem., 2011, 70, 429.
- L. Sbarcea, L. Udrescu, L. Dragan, C. Trandafirescu, Z. Szabadai and M. Bojita, Pharmazie, 2011, 66, 584.
- M. A. S. Pires, R. A. Souza dos Santos, R. D. Sinisterra, Molecules, 2011, 16, 4482.
- T. R. Sekharan, P. Sudha, S. Palanichamy and A. T. Thirupathi, Journal of Pharmacy Research, 2010, 3, 3018. 2
- C. Garnero, A. Zoppi, D. Genovese and M. Longhi, Carbohydrate Res., 2010, 345, 2550.
- L.-M. Miclea, L. Vlaia, V. Vlaia, D. I. Hadaruga, and C. Mircioiu, Farmacia (Bucharest, Romania), 2010, 58, 583.
- G. Patyi, A. Bodis, I. Antal, B. Vajna, Zs. Nagy and Gy. Marosi, J. Therm. Anal. Calorim., 2010, 102, 349.
- S. S. Shah, S. J. Pandya, T. Y. Pasha, M. K. Waghulade, K. Mahesh and P. S. Patel, Journal of Pharmacy Research, 2009, 2, 432.
- V. Mohit, G. Harshal, D. Neha, K. Vilasrao and H. Rajashree, J. Incl. Phenom. Macrocycl. Chem., 2010, 67, 39.
- N. Pravin, A. Babasaheb, D. Neha, K. Vilasrao and H. Rajashree, J. Incl. Phenom. Macrocycl. Chem., 2009, 65, 377.
- M. Jug, M. Becirevic-Lacan and S. Bengez, Drug Dev. Ind. Pharm., 2009, 35, 796.
- K. Y. Khaled, Alexandria Journal of Pharmaceutical Sciences, 2009, 23, 1.
- S. Wang, Y. Ding, Y. Y. Yanfei, Drug Dev. Ind. Pharm., 2009, 35, 808.
- W. Yang and M. M. de Villiers, American Pharmaceutical Review, 2006, 9(4), 70, 72-75.
- M. R. Caira, Mol. Pharmaceutics, 2007, 4, 310.
- M. R. Caira, S. A. Bourne, H.Samsodien, E. Engel, W. Liebenberg, N. Stieger and M. Aucamp, CrystEngComm, 2012, DOI: 10.1039/C2CE06507J.
- T. Friščič and W. Jones, J. Pharm. Pharmacol., 2010, 62, 1547.
- M. K. Stanton and A. Bak, Cryst. Growth Des., 2008, 8, 3856.
Author Biography
Mino R. Caira is Professor of Physical Chemistry at the University of Cape Town and Director of the Science Faculty’s Centre for Supramolecular Chemistry Research. His expertise is in the area of solid-state chemistry of drug polymorphs and novel multi-component systems containing APIs, especially solvates, inclusion complexes and co-crystals.
[email protected]
http://www.supramolecular.uct.ac.za/index.htm