Introduction
The release of the drug substance from the solid dosage form has a major impact on its rate and extent of absorption. In certain instances, as is the case with modified release formulations, the rate limiting step in the appearance of the drug in the systemic circulation is its release from the formulation.
In the vast majority of cases, in vitro dissolution of an immediate release product is one of the most important tools in assuring the batch-to-batch quality of the drug product. Establishing the appropriate dissolution specifications will assure that the manufacture of the dosage form is consistent and successful throughout the product’s life cycle and that each dosage unit within a batch will have the same pharmaceutical qualities that correspond to those shown to have an adequate safety and efficacy profile. Due to the critical role that dissolution plays in the bioavailability of the drug, in vitro dissolution can serve as a relevant predictor of the in vivo performance of the drug product. In this case, clinically meaningful dissolution specifications will minimize variability to the patient and therefore will optimize drug therapy.
This article discusses relevant factors that should be considered in developing a clinically relevant dissolution method and specifications. In addition, the role of dissolution in the development of the appropriate design space will be illustrated.
Choosing the right dissolution method conditions with the appropriate specifications will go a long way in assuring similar release characteristics between individual units and from lot to lot. This in turn will decrease the probability of having lots with markedly different bioavailability and therefore, inconsistent therapeutic effect.
General Principles in Selecting a Biorelevant Dissolution Method and Specification
Until recently, dissolution testing was considered to be a purely quality control tool to assure consistency from batch to batch. However, with the possibility to develop a relationship between the in vitro dissolution of a drug product and its in vivo bioavailability, dissolution testing is considered a surrogate for the in vivo performance of the drug product and is used more and more to address the impact of changes in chemistry and manufacturing controls [1,2]. In addition, biorelevant dissolution testing allows for the prediction of the in vivo performance of a drug product at least on a rank-order basis. On the other hand, products can be approved based only on the comparability of their dissolution profiles without having to conduct in vivo studies [3]. Therefore with the choice of the appropriate dissolution method and specifications, it is possible to optimize the therapeutic benefit by decreasing the variability from one lot to the other.
Considerations for the Development of an Optimal Dissolution Method
According to the FDA guidance for industry “Dissolution Testing of Immediate Release Solid Oral Dosage Form” the dissolution characteristics of the drug product should be developed considering the pH solubility profile and pKa of the drug substance [4]. The drug permeability or octanol/water partition coefficient measurement may also be useful in selecting the dissolution methodology and specifications. For NDAs, the specifications should be based on the dissolution characteristics of batches used in pivotal clinical trials and/or in confirmatory bioavailability studies. If the formulation intended for marketing differs significantly from the drug product used in pivotal clinical trials, dissolution and bioequivalence testing between the two formulations are recommended.
Dissolution testing should be carried out under mild test conditions, basket method at 50/100 rpm or paddle method at 50/75 rpm, at 15-minute intervals, to generate a dissolution profile. For rapidly dissolving products, generation of an adequate profile sampling at 5- or 10-minute intervals may be necessary. For highly soluble and rapidly dissolving drug products (BCS classes 1 and 3), a single-point dissolution test specification of NLT 85% (Q=80%) in 30 minutes or less is sufficient as a routine quality control test for batch-to-batch uniformity. For slowly dissolving or poorly water soluble drugs (BCS class 2), a two-point dissolution specification, one at 15 minutes to include a dissolution range (a dissolution window) and the other at a later point (30, 45, or 60 minutes) to ensure 85% dissolution, is recommended to characterize the quality of the product. The product is expected to comply with dissolution specifications throughout its shelf life. If the dissolution characteristics of the drug product change with time, whether or not the specifications should be altered will depend on demonstrating bioequivalence of the changed product to the original bio-batch or pivotal batch. To ensure continuous batch-to-batch equivalence of the product after scale-up and post-approval changes in the marketplace, dissolution profiles should remain comparable to those of the approved biobatch or pivotal clinical trial batch(es).
In many instances for poorly soluble drugs (BCS class II or IV), in order to obtain complete and fast dissolution of the drug product, increased amounts of surfactant, organic and/or hydro-alcoholic solutions are used as the dissolution medium in combination with relatively vigorous agitation speeds resulting in fast dissolution. Although it is possible to obtain complete dissolution of the drug from the formulation, such dissolution tests provide little value as a quality control tool because of poor discriminating ability. For such dissolution method and conditions to be acceptable and useful from a regulatory point of view, one should demonstrate the discriminating ability of the method. This can be accomplished by showing that the method can differentiate between formulations with widely different in vivo release characteristics or alternatively by showing that the method can reject lots that are not acceptable from a chemistry and manufacturing point of view. This is commonly done with drug eluting stents where the sponsors generate data to show that the dissolution method is able to reject stents with unacceptable release characteristics due to intentional manipulation of the formulations. For such a drug device combination where the intended use is over a relatively long period of time (months to years) and where the therapeutic effect cannot be easily reversed, it is crucial that the dissolution method provides the necessary quality assurance. Moreover, it becomes an extremely important tool in assessing certain chemistry and manufacturing changes since conducting in vivo bioequivalence studies in human volunteers is practically impossible.
Minimizing Variability to Obtain Consistent In-vitro Release Characteristics and Optimal Therapeutic Benefit
In the past, it was usual and customary to set dissolution specifications based on the variability in the in vitro dissolution data. The end result of this practice was the possibility of introducing lots on the market that were highly variable resulting in potentially wide fluctuations in plasma levels leading to a variable therapeutic effect and increased incidence of adverse events. Moreover, this practice of setting the limits to +/- 3 standard deviations tended to reward manufacturers with poor and highly variables formulations. Therefore, manufacturers with poor manufacturing and process controls would have products with relatively wider dissolution specifications compared to manufacturers with very tight controls in their manufacturing. Thus, the FDA is no longer accepting such a practice and it now stipulates that variability should no longer be a consideration in setting dissolution specifications. This change in policy would force drug manufacturers to tighten their manufacturing controls and to develop less variable dissolution methods.
Individual vs. Mean Performance
It has been a common practice of manufacturers to propose dissolution specifications based on passing the specifications at stage 1 of the USP acceptance criteria (all the individual units meet the specifications). This practice would result in having some units (outliers) drive the specifications. If the premise that all units should meet the acceptance criteria is accepted, this would result in dissolution specifications that would allow the release of lots with markedly different release characteristics. Such specifications would not ensure consistency from lot to lot and would not provide the best product to the patient. It is a misconception to believe that if a lot fails to meet the dissolution specification at Stage I of USP testing, this signifies that the manufacturing process is not well-controlled. In fact from a regulatory point of view, a failure exists when the lot fails to meet the acceptance criteria at stage 3 of testing. In view of the above consideration, setting the dissolution specifications based on average performance (ability to pass stage 2 testing) would result in acceptance criteria that would minimize the probability of the release of lots with atypical performance and therefore ensuring a more consistent therapeutic effect to the patient.
Assurance of Complete Dissolution of the Drug Product
The specification for the amount of drug dissolved is another important consideration in ensuring that the patient always gets the same therapeutic dose from lot to lot. For drugs that exhibit complete dissolution, setting the highest Q value possible would minimize the variability in the dose delivered to the subject. While in an ideal situation, one would like to see a Q value of 100 %, from a practical point of view this is not possible due to the fact that there is inherent variability both in the content uniformity of the dosage form and in the dissolution test. For monographs of older drugs, a Q value of greater than 75 % is seldom observed for completely dissolving drugs. However, in recent years, it is more common to see the Q value set at 80 % with some cases going up to 85 %. Such a specification would not allow the release of lots that on average differ by more than 20 % in the amount of drug delivered and thus minimizing the probability of bioinequivalence.
Appropriate Dissolution Time Specifications
While the choice of time points is clearly defined for modified release formulation in the 1997 guidance for industry, “Extended Release Oral Dosage Forms: Development, Evaluation and Application of In Vitro/In Vivo Correlations” [5], there is a debate on establishing the optimal dissolution time point for IR formulations. Most sponsors prefer setting dissolution specifications at times not faster than 30 minutes even though their product might be completely dissolved in 5 or 10 minutes. It is believed that setting a faster dissolution time specification would not translate into in vivo bioavailability differences and therefore dissolution time points faster than 30 minutes will put an undue manufacturing burden without achieving any benefit. At present it is not uncommon that both sponsors and regulators consider dissolution time point specifications as early as 15 minutes for fast dissolving formulations (100 % in less than 10 minutes). Such early time points will minimize the introduction of lots with markedly different dissolution characteristics and will ensure a more consistent performance from lot to lot.
Should All Lots Meeting Dissolution Limits be Bioequivalent (BE) to Each Other Besides Being BE to the Clinical Batch?
In an ideal situation, one would like to see that all lots allowed to be released by the specifications be bioequivalent to each other. This is not always possible because in certain cases this will constitute a heavy burden on the manufacturer and a large proportion of perfectly acceptable batches would be rejected. That is why the IVIVC guidance stipulates that at the minimum, lots that are on the upper and lower specification limit be bioequivalent to the clinical bio/lot which were used in the clinical trials and whose safety and efficacy has been established [5]. This position is deemed not acceptable by some because they believe that all batches found in the market should be bioequivalent. This is somewhat more stringent than the current practice especially for wide therapeutic index drugs. As an example let’s take 2 formulations that are bioequivalent to a clinical formulation but differing in their mean performance by 10 % on the upper and lower side of the clinical formulation. These 2 formulations most probably will not be bioequivalent to each other (since they are 20 % different on average and thus would not pass the regulatory requirement of a 90 % confidence interval of 80-125%) but will still be acceptable from a safety and efficacy profile point of view due to the fact that a 20% difference in plasma concentrations will most probably not result in any clinical difference in the pharmacological action of the drug product. Therefore, for wide therapeutic index drugs, the minimal requirement is that these lots be bioequivalent to the clinical/bio lots. This will provide regulatory relief for manufacturers without introducing into the market lots having inadequate safety and effi cacy profi les. However, for drugs exhibiting a narrow therapeutic index, the criteria should be more stringent and should require that all the lots within the dissolution specifi cations be bioequivalent to each other. It is the opinion of the author and not authors that the criteria for setting dissolution specifi cations that take into account the clinical pharmacology characteristics of the drug are more appropriate than criteria based solely on the ability to meet a statistical criterion on the plasma concentrations.
Role of Dissolution in Implementing Quality by Design (QbD) and in Defining the Design Space
Dissolution testing is a potentially powerful Critical Quality Attribute (CQA) for method development of a drug product given that it is infl uenced by many different material and process inputs (e.g., raw material particle size, compression pressure, moisture) and that it can be a predictor of in vivo drug performance. Therefore, it is commonly used as of one the endpoints in defining the design space for a given drug product. Design space in this context is defined as the multidimensional combination and interaction of input variables (material attributes) and process parameters that have been demonstrated to provide assurance of quality.
Dissolution testing plays an important role in the QbD approach. Under the QbD paradigm, dissolution testing can be used to establish a relationship between (CQA) and in vitro test methods. Thus, using a QbD approach, a design specifi cation including intended use of the procedure and performance objectives (e.g., less than 20% released at 30 min, greater that 80% at 10 h, 12-h duration), is agreed upon a priori. A structured approach, such as statistical design of experiments, is used to identify the relationship between in vivo release profiles and method conditions (medium, apparatus, sampling procedure, etc.), and the response surface (in vitro release profiles). This information is used to identify critical method parameters for controlling the release profile and the ability of the method to predict drug bioavailability. A similar procedure can be used in formulation development to identify CPIs that infl uence the release profile of the product and which can be controlled to ensure final drug product quality. If a relationship between these components is established it may be possible to waive dissolution testing in product release if other tests prove that the product specifi cations have been met (i.e. disintegration testing) and if the parameters in the design space are closely defined and monitored.
There are three possible scenarios that will be discussed below describing the role of dissolution in setting a clinically meaningful design space.
Case 1: No data relating in vitro dissolution with in vivo bioavailability.
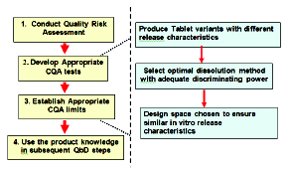
Figure 1 - Role of dissolution in defining the design. Case 1: No data relating in vitro dissolution with in vivo bioavailability
In such a case dissolution can be still used as an endpoint to define the design space. However, the dissolution method should be sensitive and discriminating to pick up difference in the critical manufacturing variable. In addition, the dissolution endpoint selected should be based on the performance of the clinical and bio lots that were shown to be safe and eff ective. The above described scenario is illustrated in Figure 1 and can be summarized as follows:
- Produce dosage forms variants with different in vitro release characteristics
- Select the optimal dissolution method that provides an adequate discriminating power
- Design Space should be chosen to ensure similar vitro release characteristics (by F2 testing or other appropriate means)
The optimal design space should contain all lots with similar release characteristics to the lots that were shown to be safe and effective. Even though the regulatory decision is made solely on in vitro considerations, the quality risk is minimized and the clinical benefi t is optimized given that no lots with different release characteristics would be allowed to be released on the market.
Case II: Established in vitro release characteristics resulting in bioequivalence.
In this scenario even though a formal predictive in vivo in vitro correlation is not established, the range of dissolution profiles or release characteristics resulting in bioequivalence are defined enabling the determination of acceptable boundaries resulting in similar in vivo performance. In this case the design space is chosen to result in bioequivalent performance within the design space.

Figure 2 - Role of dissolution in defining the design. Case II: Established in vitro release characteristics resulting in bioequivalence
Case III: Presence of in vivo in vitro correlation (IVIVC).
This is the most desirable scenario and most applicable to modified release formulations. In such a case, the rate limiting step in the appearance of the drug in the systemic circulation is its release from the dosage form. In the presence of an acceptable IVIVC model, the dissolution method is considered biorelevant allowing for the establishment of clinically relevant dissolution specifications. An acceptable IVIVC model allows for the estimation of the dissolution profile from the drug product that would be bioequivalent to the reference (a target profile for this product). The dissolution profi les predicted by the IVIVC model can be used then is setting acceptable design space boundaries that are clinically meaningful.
The general steps in setting the design space in the presence of a validated IVIVC are follows:
- Produce dosage forms variants with different in vitro release characteristics
- Select the optimal dissolution method that provides an adequate discriminating power and is predictive of the in vivo performance
- Determine the bioavailability for all the dosage form variants.
- Establish correlation between the in vitro dissolution and in vivo bioavailability (preferably a level A correlation)
- Design Space chosen based on predicted plasma concentrations that are bioequivalent to the target (clinical) formulation
In all the above three scenarios, with varying levels of assurance, dissolution is used as valuable tool to define the design space that will ensure consistent in vivo performance similar to that for the clinical trial lots.

Figure 3 - Role of dissolution in defining the design Case III: Presence of iIn vivo in vitro correlation (IVIVC)
In both scenario II and III one is able to make informed decisions on critical manufacturing variables taking into account the impact on in vivo performance. Therefore, any chosen control steps or specifi cations are tied to the clinical outcome. This is somewhat a shift in paradigm because in the past any regulatory decision with regards to chemistry and manufacturing was based solely on manufacturing capabilities and in vitro considerations. At present, dissolution is proving to be not only a valuable tool that enables an optimal quality control of the product but also a link of manufacturing considerations to linical outcomes optimizing the therapeutic benefit.
Conclusion
Dissolution is recognized as a reliable surrogate for bioavailability, therefore, it plays a major role in the ability to relate critical manufacturing parameters with clinical outcomes. A discriminating dissolution method sensitive to changes in critical manufacturing variables can be a powerful tool in establishing the design space that will minimize the quality risk to the patient. It will also allow the setting of specifi cations that are meaningful from a clinical point of view.
Even without the presence of an IVIVC, a discriminating method with the appropriate dissolution specifications would reduce the probability of having lots with different release characteristics resulting in less variability in plasma concentrations leading to a more consistent therapeutic effect and improved clinical outcome to the patient. Additionally, IVIVCs can be a powerful tool in setting clinically meaningful dissolution specifications. The ability to predict plasma concentrations from in vitro dissolution profiles will allow the setting of dissolution specifications that would ensure that all lots released would be bioequivalent to the lots that were shown to be safe and effective thus minimizing the probability of releasing lots with unproven safety and effi cacy profiles.
Acknowledgements
Dr. Sandra Suarez Sharp and Dr. Angelica Dorantes from the Biopharmaceutics group in the Office of New Drug Quality Assessment, Center for Drug Evaluation and Research, Food and Drug Administration for their valuable contributions to the article.
References
- FDA guidance for Industry: SUPAC-MR: Modified release solid oral dosage forms: scale up and post approval changes: chemistry, manufacturing and controls, in vitro dissolution testing and in vivo bioequivalence documentation 1997.
- FDA guidance for Industry: SUPAC-MR: Modified release solid oral dosage forms: scale up and post approval changes: chemistry, manufacturing and controls, in vitro dissolution testing and in vivo bioequivalence documentation 1997.
- FDA Guidance for Industry: Waiver of in vivo bioavailability and bioequivalence studies for immediate release solid oral dosage forms containing certain active moieties/active ingredients based on biopharmaceutics classifi cation system 1999.
- Guidance for Industry: Dissolution Testing of Immediate Release Solid Oral Dosage Form, August1997
- Guidance for Industry: Extended Release Solid Oral Dosage Forms: Development, Evaluation and Application of In Vitro/In Vivo Correlations, September 1997.
Author Biography
Dr. Patrick Marroum obtained his BS in Pharmacy from the University of Pittsburgh and a Ph.D. in Pharmacokinetics from the University of Florida. In 1991 he joined the FDA as a biopharmaceutics reviewer. In 1995 he became the team leader for the cardio renal team in the Offi ce of Clinical Pharmacology. In 2001 he became a regulatory science expert on in vivo in vitro correlations for modifi ed release formulations. From 2008 to September 2011, Dr. Marroum assumed the position of biopharmaceutics lead in the Offi ce of New Drug Quality Assessment where he was responsible for all the biopharmaceutics activity within the Offi ce including the review of quality by design submissions. He is presently a consultant to the pharmaceutical industry.