Tony Cundell- Microbiological Consulting, LLC, Rye, NY; Kevin L. Williams- bioMerieux, Hazelwood, MO.
Introduction
The issue at hand is not whether to include the recombinant Factor C (rFC) reagent in the U.S. Pharmacopoeia, but rather how to implement this change with minimal risk. The authors suggest that a gradual implementation approach, based on a risk analysis of the potential consequences during the transition planning phase, could help facilitate this transition smoothly. Factors that can help preclude potential risks will be explored, including sample type, examination of process microflora, reagent sample combined interference profile, pass-fail frequency examination, reagent quality awareness, and use of corroborating data.
Why should we consider a risk analysis before the adoption of the rFC reagent for bacterial endotoxin testing? The risk assessment can provide a systematic framework for the decision-making to transition from the traditional reagent (LAL) to the now well-established alternative reagent (rFC). If the benefits are self-evident, disadvantages absent or minimal, the risk of releasing a pyrogenic lot is very low, i.e., a decision equivalency of rFC to the Limulus Amebocyte Lysate (LAL)- based assay, and uncertainty around the transition low, then the U.S. Pharmacopeia should proceed with the addition of the rFC reagent to <85> Bacterial Endotoxin Tests.
The Case for rFC
The use of a sustainable recombinant protein in lieu of the harvesting of natural proteins from the horseshoe crab has begun to be implemented in earnest by many large pharmaceutical companies to provide sustainability and supply chain security advantages.1,2 The advantages accrue to the environmental conservation efforts as well regarding ensuring that supply chains are reinforced against potential reagent supply chain difficulties including (worldwide and localized) availability as well as protection against price increases.
RFC alternative validation activities have been performed ad infinitum since the original validation in 20103 with now over twelve years of hindsight the added value for each additional user-performed alternative validation is very low. Compendial status would remove the need to perform studies where the outcomes are already well known. Yes, the linearity, accuracy, and precision etc. are important but they are also readily demonstrable and have been repeated enough times to show compendial level repeatability. There is little value in its repeat performance because issues that may arise are not method related, but rather are likely relative to specific sample types and/or unique process microflora. In risk assessment, the emphasis is placed on demonstrating real-world sample-specific recovery.
Risk Analysis
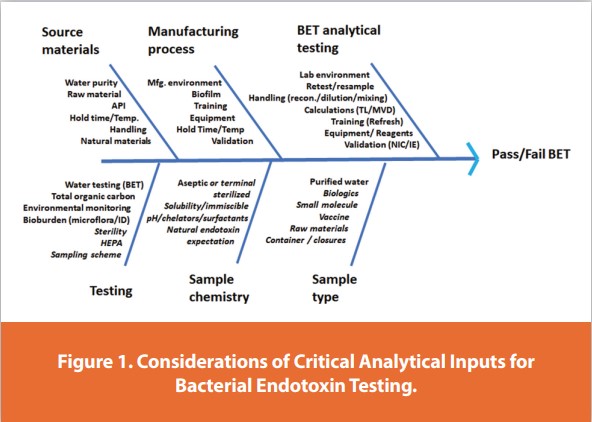
After a general overview of risk analysis relative to BET, a discussion of the major elements of risk-based decision-making will follow. Figure 1 identifies many of the critical analytical inputs for Bacterial Endotoxin Testing in a fishbone diagram.
Tables 1, 2, and 3 below assign risks in terms of probability of test failure for different sample types, likelihood of detection of bacteria endotoxin exceeding the limit, and estimated risk of severity of a contamination event.
A high-level comparison of the rabbit pyrogen and the bacterial endotoxin tests is found in Table 4.
Specific Elements to be Considered in Precluding Potential Risks
Analytical efforts that can help preclude potential risks in any risk analysis will be explored, including(i) sample type, (ii) examination of process microflora, (iii) reagent-sample combined interference profile, (iv) pass-fail frequency examination, (v) reagent quality considerations and (vi) use of corroborating data.
(i). Sample type
The introduction of a BET method, here the rFC reagent, has different impacts associated with different sample types tested. The lowest risk of the introduction of the method is with water for injection (WFI), the finished product has the highest risk in terms of impacts of failure in terms of sample complexity, cost, and patient safety. The other sample risks lie in between these two bounds. With WFI, double-distilled water would have a lower risk of failure than WFI produced by reverse osmosis. As the European Union until very recently did not authorize the use of reverse osmosis, U.S. manufacturers used distillation to sell their products globally. WFI meets strict chemical and microbiological standards and has no added substances, thus reducing the risk.
In quality testing for finished products the expectation is that the test should be performed with aliquots from beginning, middle, and end vials and the original vials should be saved for failure analysis purposes. After all, the surest indication of a lab-induced failure is a retesting of the original vials from which the original aliquots were taken. Care should be taken to aseptically remove aliquots (to pool) from those vials and to preserve those vials (minus aliquots) at the appropriate storage temperature.
Many drug companies have begun to test purified water samples routinely using rFC as a first step in gaining sustainability and supply chain assurance improvements given the ease of such a switch. The ease of change is facilitated by the lack of regulatory impediments (inspectional rather than pre-submission based), even with the current USP status as an alternative method, it is even easier with EP compendial status.
Consider that monoclonal antibody biologic products that do not have sterile downstream processes, are routinely monitored for endotoxin and bioburden. Since bioburden testing has an associated limit of NMT 10 CFU per 100 mL based on EU GMP requirements, then meeting this level ensures that the absence of febrile-level events4 provided further processing bioburden controls remain in place.
As a collective effort, we should seek common ground. The transition could be pharmaceutical-grade water in the first phase with the method published in USP <1231> Water for Pharmaceutical Use, validation requirements for non-lysate methods as published in a revised <1085> to facilitate the transition and after the development of a larger body of industry experience, add rFC reagent as an option to lysate in <85> Bacterial Endotoxin Tests after the standard enhancement/inhibition and linearity testing.
Revision to a harmonized compendial chapter will require the agreement of the U. S. Pharmacopoeia, European Pharmacopoeia, and Japanese Pharmacopoeia. At the June 2022 Pharmacopoeial Discussion Group (PDG) meeting, the Japanese Pharmacopoeia., who incidentally is designated as the coordinating pharmacopoeia for the chapter, was not ready to add the use of rFC reagent to the harmonized chapter and indicated they were sponsoring a comparative study to provide additional evidence of equivalency. It should be noted that any test sample test that is not water for Injection is a specific sample test and therefore should not impede method acceptance.
(ii) Examination of process microflora
The most critical concern of testing is the potential of the generation of false negative results in finished product testing. If there is internal concern about the detection coverage of all Gram-negative bacterial endotoxin types, an examination of the bacterial microflora types available from bioburden testing can be performed. Uncommon Gram-negative bacteria can be checked and verified in side-by-side testing with rFC and LAL reagents.
Obtaining false positive bacterial endotoxin tests, such as the detection of glucans using LAL, can be problematic in that lots of products suitable for use are rejected and the product is not available to treat patients. Also, the pharmaceutical manufacturer will experience an economic loss, see their brand impacted, and may have to contend with unhappy clinicians and their patients impacted by product supply problems. The out-of-specification investigation may possibly uncover that this is a false positive result by investigative testing with a LAL-based assay and even conducting a rabbit pyrogen test.
How can we best detect false negative and positive reactions? Analytical efforts to preempt any possibility of false negative results can focus on process microflora (available from process bioburden data). In the very rare instance of potential diminished detection of specific bacterial endotoxin in specific process settings, the previously mentioned check of any unique endotoxin types or endotoxin like PAMPs (Sphingomonas5 or Francisella6 or Heliobacter pylori) can be made to verify the detection capability via specific LAL and rFC reagents-from both a false positive and false negative perspective. Heliobacter pylori has been pointed to as a species underestimated by rFC,7 however, the likelihood of finding H. pylori in any manufacturing process is virtually nil given its fastidious growth requirements. The low reactivity of H. pylori endotoxin has been theorized to be a mechanism of persistence in human gastric ulcers.8
As part of change control requirements multiple lots may be tested in parallel with both the LAL-based and rFC-based methods. The method suitability requirements as found in USP <85> may be sufficient to detect false negative and positive reactions.
(iii) Reagent-sample combined interference profile
The product interference profile is determined relative to the Maximum Valid Dilution (MVD) of a product with a specific reagent as per USP <85> and is generated by performing a non-interfering concentration (NIC) test followed by validation close to the lowest product concentration level that overcomes interference. This is an important consideration when comparing results from different reagents that may have different abilities to overcome interference mechanisms. This includes pH, buffering capability, matrix background response including glucan cross-reactivity, and endotoxin aggregation properties. Performing tests at the same dilution with different reagents will not necessarily provide a comparison that overcomes inhibition for both tests at different dilutions. The test that retains inhibition will likely show significantly reduced values relative to the test that has overcome inhibition (a test that is neutral or shows enhancement). Some LAL manufacturers have suggested that such a comparison test should be viewed as “under detection” in the test with the lower result whereas simple method development work needs to be performed to overcome inhibition in this test. This is the purpose of the NIC testing as required in USP <85>.
(iv) Pass-fail frequency examination
To make an initial comparison, we can construct a simple pro versus cons table. Secure representative samples from a range of sample types (See Table 1) and test replicate samples using both test methods and make a statistical comparison of the total samples and the samples by sample type. Lastly, spike known amounts of bacterial endotoxin into a common sample matrix to confirm percent recovery, the limits of detection and quantification, linearity, and pass/fail rates around a bacterial endotoxin limit for a range of products. The major advantages of the rFC reagent are its expected greater lot-to-lot consistency, non reliance on an animal source, and insensitivity to cellulose-derived glucans. When using highly sensitive assays, there may be a statistical difference between the numerical results obtained using the various methods when analyzing the same sample, but there will be no difference in the frequency with which the bacterial endotoxin limit for the same product will be passed or failed. As recommended in USP <1223>, the results should be comparable with a non-inferiority test applied to the data.9 Other critical issues are the breadth of sample type evaluated and the number of samples tested to provide sufficient statistical power to the analysis.
(v). Reagent quality considerations
The LAL reagent is a biological product licensed by the Center for Biologics Evaluation and Research (CBER), Food and Drug Administration purified from the hemolymph of the North American Horseshoe Crab, Limulus polyphemus, whereas recombinant Factor C (rFC), is a biotechnology-manufactured version of the same Factor C protein that is the triggering LAL biosensor that does not require licensing by CBER or CDER. Some in our industry believe this difference in regulatory oversight increases the routine test risk. However, LAL reagent has been historically regulated due to its status as a blood derived product, it is the only compendial test reagent so regulated.10 Although not regulated, as any other biomanufacturing process, the rFC production undergoes drastic quality control checks to assess its quality along the process. Drug companies can audit any reagent manufacturer as well as track various reagent performance characteristics over time as reagent lots change. For example, reaction times for a BET may change significantly from batch to batch and users should monitor these changes. For fluorescent methods, any change in the reagent batch will trigger a new gain test to ensure continued optimal performance [delta relative fluorescence units(dRFU’s)].
(vi) Use of corroborating data
Corroborating data may include the use of bioburden data and associated bacterial identification to verify that unusual flora does not present false positive or false negative possibilities within the process flow.
Another set of corroborating data is associated with purified water testing for Total Organic Carbon (TOC). TOC while not directly correlated with bacterial endotoxin, an increase can be viewed as an indicator of bacterial growth in the form of accumulating biofilm.
Experience with the Transition from the Rabbit Pyrogen Test to the LAL-Bacterial Endotoxin Test
What can be learned from the transition from the Rabbit Pyrogen Test to the LAL-Bacterial Endotoxin Test? The history of this transition is well described in the literature.11 The Rabbit Pyrogen Test was used for over 30 years to test and release pharmaceutical and biological products. The challenges were vastly greater in this transition than in the current transition. They included manufacturing the reagent, establishing reference and control standards, standardizing the tests, establishing the threshold pyrogenic level for bacterial endotoxin, developing a regulatory framework, implementing a globally recognized reference standard, setting bacterial endotoxin limits for different parenteral products, and demonstrating equivalency of the two methods. The LAL to rFC transition is less challenging and may eventually be viewed as a like-to-like reagent substitution.
Patient Safety Risk from Parenteral Drug and Medical Devices Contaminated with Bacterial Endotoxin
A recent review article12 addressing the period of 2011 to 2021 using FDA data from Good Manufacturing Practice non-compliance observations, product recalls and the Adverse Event Reporting System clearly demonstrated an absence of industry issues with bacterial endotoxin. For example, with the 188 GMP compliance observations reported, 70% and 30% were related to laboratory testing and manufacturing respectively with 56% associated with finished drug product testing. In contrast, 95% of the endotoxin related recalls were associated with medical devices. These findings reinforce the effectiveness of the Bacterial Endotoxin-LAL assay. All current indications are that rFC will present a continuation of this well established safety record.
Potentially Confounding Issues
Another minor risk associated with the implementation of the rFC based assay is an upward or downward shift in the historic bacterial endotoxin results so adverse trending rules may need to be reset. It is recognized that testing within the MVD will confirm that the lot does not exceed the bacterial endotoxin limit but to quantify the bacterial endotoxin level test should be conducted at the lowest possible dilution to overcome inhibition or enhancement of the assay (as per 2012 FDA Q&A Guideline). Many laboratories routinely select a dilution 2- or 10-fold lower than the MVD when testing the product and trending the results.

Confounding issues associated with BET testing in general include differences found in naturally occurring endotoxin and highly purified reference standard endotoxin. It also includes enhancement of results by glucans in test materials tested by LAL (variable as affected by various LAL formulations), the simplification due to the application of the concept of the threshold pyrogenic effect (using purified LPS), the assumption that endotoxin is homogeneously distributed throughout a product, the presence of non-endotoxin febrile responses in patients (routine for some immune modulating biologics), and the non-adherence of clinicians to the recommended dosage when administering the drug (see gentamicin episode).13
Frequency of Testing and Sample Type
It is estimated that over 70 million Bacterial Endotoxin Tests are conducted globally annually. As an estimated 90% plus of the reagents sold are animal-derived, the transition could relieve potential disruptions to the bacterial endotoxin testing market if the LAL reagent comes to be in short supply.
It is revealing to explore the percentage of Bacterial Endotoxin Tests in support of pharmaceutical-grade water, pharmaceutical ingredients, cell culture media, buffers, in-process samples, and finished products during the manufacture of three major industrial manufacturing sectors – small molecules, large molecules, and cellular therapies. Rough estimates made by the authors are found in Table 5.
Review of the Existing Literature Supporting the Transition from the LAL to the rFC-based Bacterial Endotoxin Test
A comprehensive review of the peer-reviewed literature on the equivalency of LAL and rFC-based Bacterial Endotoxins Test supports the transition.14
Conclusions
Recombinant Factor C (RFC) reagent use has the potential to bring positive new dimensions to the current BET test paradigm including test modernization, increased supply chain security and sustainability in support of the horseshoe crab as a keystone species as well other dependent species (such as shorebirds that need the HSC egg mass as foodstuff for their rigorous migratory journeys).
Based on risk analysis principles and a review of the published literature, the authors will be interested to see the actions taken by the new USP Microbiology Expert Committee. The overview of stepwise risk assessment considerations and potential preclusion exercises proposed here are intended to support those discussions and decisions.
References
1. Guest, M., Duncanson, P., Capper, K., & Wong, D. (2022). A strategic approach to optimisations of testing bacterial endotoxins. Euro. Pharma. Rev., 24 August
2. Sustainability Bond Allocation and Impact Report, Eli Lilly & Co., 2021.
3. Loverock B, Simon B, Burgenson A, Baines A. (2010). A recombinant factor C procedure for the detection of Gram-negative bacterial endotoxin. Pharm. Forum, 36:321–329
4. Microbial Monitoring for Biological Drug Substance Manufacturing: An Industry Perspective. Corresponding author: David Bain, Facilitator, BPOG Corresponding author contact information: BPOG, 5 Westbrook Court, Sharrow Vale Road, Sheffield, S11 8YZ, United Kingdom, [email protected]. Published in a ‘Special Section’ of the PDA J. Pharm. Sci & Technol, in May/June 2015.
5. Shahar, E., Emquies, K., Bloch, I. et al. (2023) Endotoxin-free gram-negative bacterium as a system for production and secretion of recombinant proteins. Appl. Microbiol. Biotechnol. 107:287–298
6. Okan, N. A. and D. L. Kasper (2013) The atypical lipopolysaccharide of Francisella, Carbohydr. Res. 378: 79–83.
7. Stevens, I et al (2022) Advanced recombinant cascade reagent PyroSmart NextGen® for bacterial endotoxin test as described in the pharmacopeias, BPB Reports 5:105-114
8. Leker, K., Lozano-Pope, I., Bandyopadhyay, K. et al. Comparison of lipopolysaccharides composition of two different strains of Helicobacter pylori . BMC Microbiol. 17, 226
9. Validation Strategy for New Recombinant Factor C Users, Evelyn Der, Senior Scientist, Roche Genentech, QC Technology Innovation and Implementation Group, South San Francisco, CA, USA. Carmen Marin, QC Scientist Microbiology, F. Hoffmann La Roche Ltd, Kaiseraugst, Switzerland. Viviane Grunert da Fonseca, PhD, Non-clinical Statistician, Roche, MSAT (Manufacturing, Science and Technology), Penzberg, Germany. Lindsey Silva, PhD, QC Director, Roche Genentech, Microbiology, South San Francisco, CA USA., Amer. Pharm. Rev., Feb. 28, 2022.
10. Berzofsky, R.N. (2004) Does Endotoxin Testing = FDA Licensing? Cambrex (now Lonza), vol. 104, issue no. 1
11. Cooper, J. F. 2011 Discovery and acceptance of the bacterial endotoxins test In The Bacterial Endotoxins Test – A Practical Guide edited by Karen Z. McCullough DHI/PDA pp1-14
12. Tidswell, E. C. (2023) A non-trivial analysis of patient safety risk from parenteral drug-and medical device-borne endotoxin. Drugs in R & D 23:65-76.
13. Endotoxin-Like Reactions Associated with Intravenous Gentamicin -- California, 1998, MMWR, October 23, 1998 / 47(41):877-880. https://www.cdc.gov/mmwr/preview/ mmwrhtml/00055322.htm#:~:text=Parenteral%20antimicrobials%20such%20as%20 gentamicin,effects%20when%20infused%20into%20humans.
14. Bolden, J., et al (2020) Current available recombinant alternatives to horseshoe crab lysates: Are they comparable for the detection of environmental endotoxins? A review. PDA J. Pharm. Sci. & Technol. 74 (5): 602-611
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special
offers from American Pharmaceutical Review delivered to your inbox!
Sign up now!