Introduction
Understanding the biopharmaceutical properties of drug substances has become crucial for different phases of pharmaceutical research and development. One of the major goals during research phases such as lead optimization is to design molecules to allow sufficient absorption of the drug. Therefore, medicinal chemists usually choose an iterative approach to improve drug properties such as solubility, lipophilicity and permeability while maintaining and also improving properties such as potency, selectivity, metabolism and toxicology. Nevertheless the above mentioned physicochemical parameters just represent surrogates which are much easier to measure compared to assess in-vivo absorption. To predict absorption from physicochemical parameters a set of different approaches has been developed during the last decades. These include concepts such as the well-known “biopharmaceutical classification scheme (BCS)” [1], which has been refined during recent years [2], or the concept of “maximum absorbable dose (MAD)” [3]. More recently computer simulations with physiologically based pharmacokinetic absorption models have been introduced [4-7]. Therefore, commercial software such as GastroPlus, SimCyp and PKSim today is available. Most of the approaches used within these tools are based on the “compartmental absorption and transit model (CAT)” [8] and further developments of this. Amongst other parameters, the CAT takes into account physicochemical properties of the drug molecule, calculates concentrations of the drug in the distinct parts of the gastrointestinal tract and determines absorption of the drug mainly into the portal vein. Actual concentrations of the drug refer to different states of the molecule such as the dissolved neutral molecule and also charged (e.g. protonated or deprotonated) species. Furthermore, several undissolved species such as parent (free base, free acid) and salt forms are taken into account, and inter-conversion between these forms is accounted for in the pharmacokinetic simulation (PK simulation). The respective conversion rates are governed by the whole set of physicochemical properties of the drug molecule as well as the physiology used for the model (e.g. mouse, rat, dog, human). As the latter is independent from the drug molecule, we focus the further discussion on the physicochemical parameters of the drug.
The scope of the discussion will be to show how physicochemical parameters of drugs can be efficiently generated and used to understand drug absorption based on simulation of absorption processes by PK simulation during pharmaceutical research.
Setting the scene: physicochemical parameters governing drug absorption
Absorption of drugs from solid oral dosages is mainly governed by three processes: (i) liberation of the drug particles from the dosage form, (ii) solubilization of the drug in the gastric or intestinal fluid, and (iii) permeation of the molecular dispersed drug through the gut membrane. Bioavailability of drugs from solid-oral dosage forms is nowadays often limited by either the solubilization process (reflecting the trend towards poorly soluble drugs), or insufficient permeability (e.g. peptide-based drugs).
Solubilization Process
Solubilization of drug molecules in the gastro-intestinal (GI) tract is driven by both equilibrium solubility, as well as the kinetics of the dissolution process, since residence time in each gastrointestinal compartment is limited. Whereas thermodynamic solubility is essentially an intrinsic property of the drug, dissolution kinetics strongly depends on the chosen solid-state form, as aspects such as salt entity [9], (pseudo)-polymorphic form [10] as well as particle properties (size, shape, surface area, surface polarity) strongly impact on the dissolution behavior.
Solubility and dissolution studies under variation of pH are essential in-vitro experiments to mimic behavior under physiological conditions. Most drugs exhibit strong pH-dependence on these parameters if drug ionisation constants (pKa) are in the physiological relevant pH-range 1-7. This may also be accompanied by a change in the respective solid-state form, as covered by the concept of pH-max [11]. This concept describes at which pH solubility will be governed by solubility of the parent and at which pH solubility of a salt form becomes crucial. The concept of pH-dependent solubility and dissolution behavior was further progressed to mimic biorelevant media, with media like FaSSIF and FeSSIF being introduced [12-14]. Whilst the conventional approaches to study solubility and dissolution behavior in classical pH-buffer systems tend to deliver underestimated figures for these parameters, the scope of the biorelevant media approach is to also reflect the solubilization effect of in-vivo relevant surfactants such as bile salts and lecithine as well as prediction of potential food-effects resulting from this.
Permeation Process
Membrane permeation of solubilized drugs may occur via multiple mechanisms, such as passive diffusion using the transcellulare or paracellulare route or active transporter mediated permeation.
Although more sophisticated in-vitro permeation tests such as PAMPA or Caco2 assays are nowadays well-established to directly measure the dynamics of effective intestinal permeation rate Peff [15], in-vitro partition coefficients (log P) and distribution coefficients (log D) represent readily obtainable physical parameters which have been shown to be suitable surrogates for estimation of intestinal permeability [16]. Although high lipophilicity in principal favors drug permeation, it is a key challenge for every drug designer to find an optimum balance between overall drug lipophilicity and hydrophilicity [17]. Pronounced lipohilicity will not only limit aqueous solubility and dissolution kinetics as a consequence of poor wettability, but may also bear the risk of membrane retention.
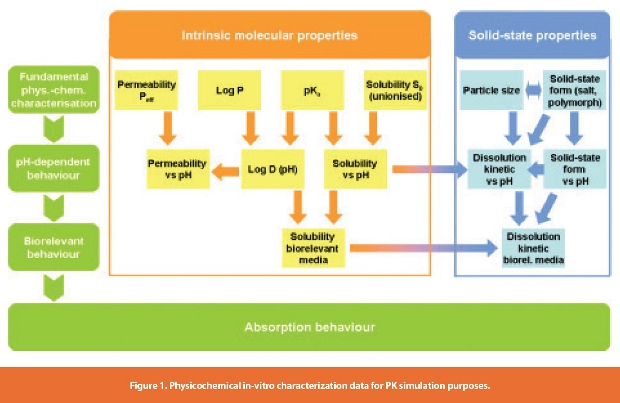
In the context of lipophilic versus hydrophilic balance, an important aspect is the amphiphilic character of a drug. In-vitro determination of drug amphiphilicity via surface tension measurements could directly be correlated to drug blood-brain barrier penetration of poorly soluble drugs [18]. A schematic representation of physicochemical in-vitro characterization data, which are relevant for PK simulation purposes, is displayed in Figure 1.
Getting started: physicochemical parameters from in-silico tools
For most physicochemical parameters in-silico predictions can be used as a first guess. Especially for lipophilicity (via log P and log D), solubility, and pKa, in-silico assessment has been introduced for a long time [19, 20]. Software tools for the prediction of these properties typically are developed using a set of compounds for which the respective property has been measured as training set. Nevertheless one has to be aware of the accuracy of these in-silico results. From our experience, most software packages will be able to calculate log P within +/-1 for the experimental result. Differences between experimental and measured results typically will be higher for log D as most software packages calculate log D from log P and pKa. While in-silico results for pKa are usually highly reliable, large deviations of several units from experimental results may also be encountered. This is often the case if the software misinterprets a certain functional group or struggles with tautomeric forms. Therefore, in-silico results for pKa should always be carefully checked for plausibility. Differences between experimental and calculated solubility can vary typically within a factor of 50. For solubility it has to be taken into account that for solid oral formulations thermodynamic solubility represents the key driver, as this property answers the question “to which extent does the compound dissolve?" Kinetic solubility starting from the predissolved compound can be misleading for this purpose as this refers to precipitation of a compound [21]. Thermodynamic solubility is controlled by hydration energy and lattice energy of the respective compound. The combination of these two terms makes calculation of solubility still today a challenging task. Prediction of solubility gets even more complicated as soon as biorelevant media such as FaSSIF and FeSSIF are considered as for this also interaction with bile salts forming micellar structures becomes relevant.
Getting better: physicochemical parameters from in-vitro tools
As soon as solid material is available, in-vitro measurements of compound physical properties should be envisaged to refine PK simulations.
Solubilization
Solubility and dissolution kinetics can reliably be accessed experimentally with in-vitro assays in small scale down to few mg, preferable 1-2 mg of compound per condition tested. As usually more than one condition (e.g. medium, pH-value) is of interest for PK simulation studies, sample quantities of ~10-50 mg are from our experiences required to compile a high-quality in-vitro data set for these parameters.
Solubility refers to thermodynamic solubility at a given condition. Although kinetic solubility assays from DMSO stock solutions are widely established mainly in early-stage characterization [21], this tends to significantly overestimate solubility of a drug in a given medium [22], e.g. due to super-saturation effects and/or amorphous precipitates. Therefore, thermodynamic solubility via shake-flask method should be envisaged, which can be considered the gold standard among in-vitro assay for solubility. The assay comprises three steps:
- Equilibration of an excess quantity of powder sample in a given medium (e.g. buffer) until equilibrium between solid-state and solution phase is established (typically 24 hours)
- Solid-liquid separation, via filtration or centrifugation as methods of choice: Whereas filtration (typically via syringe filter membranes with 0.2-0.45 μm pores) may underestimate solubility due to filter adsorption effects, centrifugation tends to result in overestimation of solubility if fine particles are not sufficiently separated from the supernatant.
- Determination of concentration level of dissolved drug in the liquid phase, which should ideally be done by HPLC to allow for detection of potential degradation products caused by the solubility assay.
Dissolution kinetics refers to the dynamics of the dissolution process over time at a given condition. In the simplest set-up, this can be studied in a beaker experiment by stirring excess quantities of powder sample in a given medium, withdrawing defined aliquots of dispersion at defined timepoints, and measure concentration levels of dissolved drug in the solution after solid-liquid separation. To allow for a defined and reproducible set-up, which is crucial in dissolution data acquisition, ideally dedicated dissolution testing systems as described in Ph. Eur. (section 2.9.3) and USP (General Chapter <711>) are being used. During early stages also miniaturized setups of this assay requiring only about 50 - 100 mg can be helpful. For improved in-vitro-in-vivo-correlations (IVIVC), the use of Flow-Through-Cell systems (USP-4) has gained wide acceptance [23], mainly due to the improved simulation of hydrodynamics, optimal sink-conditions being realized, and elimination of dead zones and coning effects which are often observed in basket or paddle dissolution set-up.
Dissolution data are strongly impacted by the solid-state properties of the drug. Therefore, it is recommended to study dissolution behavior on more than a single batch.
Permeability
Besides solubility, permeability is a key parameter governing drug absorption. PK simulation tools describe absorption either by partition coefficients or permeability as obtained from CaCo2 or similar assays. Partition and distribution coefficients can be obtained as rough starting points from in-silico models. The next step to get a better measure of drug permeability is given by partition coefficients such as log P and log D which can be obtained from simple in-vitro assays. Log P – the water-octanol partition coefficient of the uncharged drug molecule – can easily obtained e.g. from methods based on HPLC retention time. As the charge of the molecule – especially in the case of weak bases and acids – can vary over the physiological pH range in the intestine, it is crucial to know partition coefficients as a function of pH. This data set can be gathered by measuring log D at several pH values. Partition coefficients expressed as log D which refers to the distribution of the total amount of the drug regardless of its charge can easily measured by shake flask assays. Instead of doing this at several pH values using different buffer systems, it is generally simpler and more common to determine log D experimentally at a single pH and calculate log D as a function of pH using the pKa value of the drug. Beyond pH-dependence of solubility, this is the second point where acid-base constants come into play for modelling of absorption. In terms of compound consumption, all these physicochemical assays relevant for permeability require only a few milligrams of the compound. Therefore it is usually possible to obtain the respective results quite early during pharmaceutical research.
Still the same holds true for permeability as obtained from assays which mimic intestinal epithelium cells (e.g. CaCo2) in humans and accordingly are closer to in-vivo situations. Permeability as obtained from in-vitro tests represents a reliable model for drug absorption, due to activity of drug transporters which better mimic the in-vivo situation.
Particle Size
Particle size represents an important parameter which controls dissolution. Accordingly its role becomes important, where no dissolution data is available. In these cases dissolution rate can be described by the Noyes-Whitney equation: dW/dt = dissolution rate; A = surface of solid = 4π(d/2)2, d = particle diameter, C = actual concentration of compound in solution, CS = saturation concentration of compound in solution, D = diffusion coefficient, L = thickness of diffusion layer

As a first starting point for particle diameter as required for calculating dissolution rate by this model, particle size can be obtained from microscopic methods such as optical microscopy or scanning-electron microscopy. The big advantage of these methods is that they require just a few milligrams of the compound. This will be available also for small batches of compounds used for early PK studies in animals. One disadvantage of the microscopic methods is that typically they only allow a rough estimate for particle size and only a mean value is obtained – all particles are assumed to have the same diameter. At least this holds true unless the microscopic methods are combined with image analysis tools delivering particle size distributions. As soon as batch size becomes sufficient – particle size can be obtained from methods such as laser diffraction yielding true particle size distributions which might be mono- or multi-modal. During PK simulations, most of the PK simulation tools treat these distributions by erosion based on the Noyes-Whitney equation.
Conclusion
The in-vivo behaviour of new chemical entities needs to be estimated and understood as early as possible in pharmaceutical research and development. Therefore, during the last years software tools to calculate and understand pharmacokinetics including drug absorption have been introduced as valuable tools. These tools rely heavily on physicochemical parameters of drugs. In this contribution we have summarized useful ways to gather the required set of physicochemical data.
References
1. G.L. Amidon, H. Lennernas, V.P. Shah, J.R. Crison, “A theoretical basis for a biopharmaceutic drug classification: The correlation of in-vitro drug product dissolution and in-vivo bioavailability”, Pharm. Res. 1995, 12, 413-420.
2. J.M. Buttler, J. Dressman, “The developability classification system: Application of biopharmaceutics concepts to formulation development”, J. Pharm. Sci. 2010, 99, 4940-4954.
3. W. Curatolo, “Physical chemical properties of oral drug candidates in the discovery and exploratory development settings” Pharm. Sci. Technol. Today 1998, 1, 387-393.
4. J. Yokoe, N. Iwasaki, S. Haruta, K. Kadano, K. Ogawara, K. Higaki, T. Kimura, “Analysis and prediction of absorption behavior of colon-targeted prodrug in rats by GI-transit-absorption Model”, J. Control. Release 2003, 86, 305-313.
5. B. Agoram, W.S. Woltosz, M.B. Bolger, “Predicting the impact of physiological and biochemical processes on oral drug bioavailability”, Adv. Drug. Deliv. Rev. 2001, 50, 41-67.
6. D.A. Norris, G.D. Leesman, P.J. Sinko, G.M. Grass, “Development and predictive pharmacokinetic simulation models for drug discovery”, J. Control. Release 2000, 65, 55-62.
7. Y. Plusquellec, C. Efthymiopoulos, P. Duthil, G. Houin, “A pharmacokinetic model for multiple sites discontinuous gastrointestinal absorption“, Med. Eng. Phys. 1999, 21, 525-532.
8. L.X. Yu, J.R. Crison, G.L. Amidon, “Compartemental transit and dispersion model analysis of small intestinal transit flow in humans”, Int. J. Pharm 1996, 140, 111-118.
9. S. Li, S. Wong, S. Sethia, H. Almoazen, Y. M. Joshi. A. T. M. Serajuddin, “Investigation of Solubility and Dissolution of a Free Base and Two Different Salt Forms as a Function of pH”, Pharm. Res. 2005, 22, 628-635.
10. P. Lehto, J. Aaltonen, M. Tenho, J. Rantanen, J. Hirvonen, V. P. Tanninen, L. Peltonen, „ Solvent- Mediated Solid Phase Transformations of Carbamazepine: Effects of Simulated Intestinal Fluid and Fasted State Simulated Intestinal Fluid “, J. Pharm Sci. 2009, 98, 985-996.
11. A. T. M. Serajuddin, “Salt Formation to Improve Solubility”, Adv. Drug Del. Rev. 2007, 59, 603–616.
12. J. B. Dressman, G. L. Amidon, C. Reppas, V. P. Shah, Pharm. Res., “Dissolution testing as a prognostic tool for oral drug absorption: immediate release dosage forms” 1998, 15, 11-22.
13. E. Galia, E. Nicolaides, D. Horter, R. Lobenberg, C. Reppas, J. B. Dressman, “Evaluation of various dissolution media for predicting in vivo performance of class I and II drugs”, Pharm. Res. 1998, 15, 698-705.
14. E. Jantratid, N. Janssen, C. Reppas, J. B. Dressman, “Dissolution media simulating conditions in the proximal human gastrointestinal tract: an update”, Pharm Res. 2008, 25, 1663-1676.
15. A. Avdeef, “High-throughput Measurement of Permeability Profiles”, in: “Drug Bioavailability: Estimation of Solubility, Permeability, Absorption and Bioavailability”, Wiley-VCH 2004, 46-60.
16. S. Winiwarter, N. M. Bonham, F. Ax., A. Hallberg., H. Lennernas, A. Karlen,, „Correlation of human jejunal permeability (in vivo) of drugs with experimentally and theoretically derived parameters. A multivariate data analysis approach, J. Med. Chem. 1998, 41, 4939-4949.
17. C. A. Lipinski, “Drug-like Properties and the Causes of Poor Solubility and Poor Permeability“, J. Pharm. Tox. Meth. 2000, 44, 235-249.
18. A. C. Petereit, , K. Swinney, J. Mensch, , C. Mackie, S. Stokbroekx, M. Brewster, J. B. Dressman, “Prediction of blood-brain barrier penetration of poorly soluble drug candidates using surface activity profiling”, Eur. J. Pharm. Biopharm. 2010, 75, 405-410.
19. A.Klamt, B.J. Smith, „Challenge of drug solubility prediction“ in „Molecular Drug Properties“, VCH Weinheim 2008, 283-306.
20. R.Manhold, C. Ostermann, “Prediction of log P with substructure-based methods”, in „Molecular Drug Properties“, VCH Weinheim 2008, 357-380.
21. A.C. Petereit, C. Saal, „What is the solubility of my research compound?”, Am. Pharm. Rev. 2011, 14, 70-73.
22. B. Hoellke, M. Arlt, C. Saal, „Comparison of Nephelometric, UV-Spectroscopic and HPLC Methods for High-Throughput Determination of Aqueous Drug Solubility in Microtiter Plates“, Analytical Chemistry, 2009, 81, 3165-3172.
23. C. Schroter, C Wahling, A. Hanefeld, „Flow-Through Cell Method and IVIVR for Poorly Soluble Drugs“, Diss. Tech., 2011, 11, 15-24
Author Biographies
Christoph Saal, Ph.D. studied chemistry at the Technical University of Darmstadt and graduated in Physical Chemistry. In 1999, he joined Merck KGaA where worked in Central Analytics and Medicinal Chemistry. Currently, Christoph Saal is heading a group focused on analytical and physico-chemical characterization of New Chemical Entities.
Anna Christine Petereit, Ph.D. has studied Pharmacy at the Goethe University of Frankfurt and will finished her Ph.D. in Pharmaceutical Technology targeting the prediction of oral bioavailability based on surface activity profiling. She has further experience in formulating solid oral dosage forms. Currently Anna works as a Scientist at Merck KGaA in the division for physical-chemistry properties characterization with the main focus on solubility and dissolution.
Axel Becker graduated in Analytical Chemistry at Reutlingen University of Applied Sciences in 2000 and joined Merck KGaA in 2001. At Central Analytical Services, he is strongly involved in solid-state selection and physical properties characterization of research compounds and development candidates. From 2005 to 2007, Axel spent a two-year secondment at Merck Generics UK, working on physical properties characterization and Intellectual Property aspects of Generic Drugs.