Introduction
The Goal of the Track
Over recent decades, solubility has become one of the key parameters in pharmaceutical research and development as it represents a key prerequisite for ensuring adequate bioavailability of a compound [1, 2, 3, 4]. Due to different factors, such as the increasing importance of combinatorial chemistry in the 1990s and the start of the new millennium, high-throughput screening (HTS) and growing interest in targets with more lipophilic binding sites, the solubility of research compounds has become a limiting parameter in many cases [5, 6]. Accordingly, after the definition of a certain starting point represented, for example, by a HTS hit or series of hits or a starting point defined by nature, such as an endogenous ligand, drug likeness needs to be improved [7]. These starting points frequently only deliver activity in a biochemical or cellular assay. Drug-like parameters, including solubility, are consequently optimized by medicinal chemists during hit to lead optimization phases. A toolbox of structural modifications, such as introduction of hydroxyl groups, aliphatic amines rendering basicity or carboxylic groups rendering acidity, can therefore be used [8].
As solubility is optimized, it has to be born in mind that optimization of solubility is important for achieving different aims. Depending on the goal, attention has to be paid to different aspects of the solubility measurement and also the research compound. Depending on the desired aim, both measurement and consequences for the compound may be easier to handle or more challenging. Goals for optimization of solubility might be:
- design of HTS-screening libraries with sufficient solubility
- assessment and optimization of the solubility of research compounds to ensure that these compounds dissolve in various assay formats
- assessment and optimization of the solubility of research compounds for pharmacokinetic (PK) and pharmacodynamic (PD) animal studies
- assessment and optimization of the solubility of research compounds for toxicological studies
- assessment and optimization of the solubility of research compounds which are intended as development candidates for clinical trials
These different objectives define which parameters have to be addressed in connection with solubility. The easiest question might be to ensure that a research compound possesses sufficient solubility in a certain assay format. The basic aim is to investigate whether a compound precipitates in the particular assay. As most assays – such as biochemical or cellular assays, buffer, plasma or microsomal stability, PAMPA or CaCo2 assays – are carried out using compounds predissolved in DMSO solutions, the method for assessing solubility has to focus on kinetic solubility. Usually, the research compound dissolved in DMSO is pipetted into the particular buffer used in the assay. After a phase separation step, such as centrifugation or filtration, the concentration of the dissolved compound is measured by HPLC or UV spectroscopy [9]. Alternatively, it is only checked whether the compound precipitates at the desired concentration or not. This can easily be carried out by nephelometry [10].
Beyond kinetic solubility – as discussed above – solubility becomes a more complicated parameter as we move towards thermodynamic solubility. In contrast to kinetic solubility, the term thermodynamic solubility does not refer to the question “does the predissolved compound precipitate if introduced into an aqueous buffer system – and if so, what concentration is reached?” but to the question “does the solid compound dissolve if introduced into an aqueous buffer system – and if so, what concentration is reached?” In contrast to kinetic solubility, where the physical state of the compound is precisely defined as a solution in an organic solvent, for thermodynamic solubility the physical state of the compound might vary. The solid compound may exist – even if the salt form is clearly defined - in different polymorphic or pseudopolymorphic forms (solvates or hydrates) or may be amorphous. Thermodynamic solubility will be dependent on the respective solid state form. As thermodynamic solubility is defined as the concentration of a compound in equilibrium with the solid residue, it refers to the respective solid state form. Thermodynamic solubilities for different polymorphic and pseudopolymoprhic forms typically vary within a factor of 1–5, whereas amorphous phases of the same compound frequently show thermodynamic solubility one or two orders of magnitude higher [11].
Accordingly, metastable polymorphic and pseudopolymorphic forms and amorphous phases have on the one hand the chance of enhanced solubility and bioavailability, but on the other hand are also associated with the risk that the respective metastable form might not be stabilized sufficiently. As a consequence, the thermodynamically stable polymorph is typically selected for clinical development of new chemical entities. Metastable or amorphous phases might only be appropriate for challenging development candidates using suitable formulation techniques for stabilization of the drug product, such as for example hot melt extrusion. The situation could become even more challenging as it might happen that complete information about polymorphs is not available at the beginning of development, as was the case for Ritonavir [12] and Telmisartan [13].
Compared with the aspects of solubility discussed above for screening or characterization assays and clinical development, in-vivo experiments such as PK, PK/PD and toxicological studies represent an intermediate situation, where it has not yet been necessary to have a solid state form which is stable over a shelf life of several years and metastable solid state forms may be useful for achieving higher solubility. Nevertheless, care has to be taken to ensure that the behavior of the research compound is reproducible, which means that the solid state form should be controlled, for example making sure that PK/PD studies are always carried out using the amorphous phase.
Aspects to Consider When Attempting to Improve Solubility
Following the discussion above, the definition of the chemical structure and solid state form of a compound entering clinical development clearly represents a crucial milestone. As chemical structures are modified by medicinal chemists in the lead optimization phase in order to optimize compound properties, including solubility, it must be ensured that this is carried out in the right direction. Bearing in mind that, except for special cases, a crystalline phase will be selected for clinical development, it must be ensured that solubility data used for optimization of the solubility refer to the crystalline state. Otherwise, compounds belonging to a certain series might be developed not in the direction of improving their thermodynamic solubility, but instead in the direction of making crystallization more difficult. This would lead to the situation that, on proceeding to clinical development, there will come a moment when it is recognized that the solubility does not refer to a crystalline residue, but to an amorphous one. The compound will consequently have to be changed to a crystalline phase. This will lead to a considerable drop in solubility – the signpost where it would be realized that the wrong track had been followed.
Old Fashioned and Modern Signposts
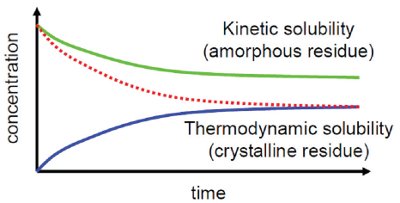
Figure 1 - Comparison of thermodynamic and kinetic solubility. Whereas thermodynamic solubility provides and answer to the question of how much a compound dissolves (blue line), kinetic solubility answers the question to which extent a compound precipitates and how much of it stays in solution. In many cases kinetic solubility is higher compared to thermodynamic solubility as precipitates typically are amorphous (green line) and not crystalline (dotted red line).
In terms of throughput, ease of handling, automation and consumption of compound, the use of kinetic solubility results as signposts for improving solubility might be seductive. However, it must be remembered that the only thing that can be learned from the determination of kinetic solubility is the answer to the question “does it precipitate?” At first glance this might not seem particularly disadvantageous, as in the equilibrium state answers to the question “up to which concentration does it precipitate?” and “up to which concentration does it dissolve?” should be identical. As can be seen in Figure 1, curves representing dissolution (blue line) and precipitation (dotted red line) ultimately converge. Nevertheless, pipetting a compound predissolved in an organic solvent such as DMSO into an aqueous buffer system, as during the determination of kinetic solubility, typically produces an amorphous precipitate [9]. Accordingly, kinetic solubility usually refers to the amorphous phase. Results of kinetic solubility measurements are typically higher than the results obtained from the same compounds by measurement of thermodynamic solubility. It can be seen that the precipitation of the compound as shown by the green line in Figure 1 does not approach the thermodynamic solubility, but remains on a higher level. Accordingly, determination of the kinetic solubility may be highly misleading for compound optimization in the direction of a clinical development candidate.
Consequently, determination of the thermodynamic solubility represents a more appropriate signpost for finding the right way to improve compound solubility. Methods for determining thermodynamic solubility typically start by incubation of the solid research compound together with the respective buffer system for a certain time (for example 24 hours) in order to ensure that equilibrium is reached between the solid and liquid phases. The solid and liquid phases are then separated by filtration or centrifugation, and the concentration in the liquid phase is determined in a similar way to the determination of kinetic solubility by HPLC or UV spectroscopy [14].
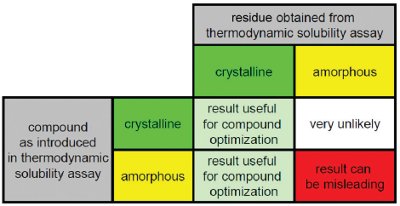
Figure 2 - Scenarios one can encounter during incubation of a research compound in an aqueous buffer system. Depending on whether the research compound is introduced as crystalline or amorphous material in the thermodynamic solubility assay, results obtained from solubility measurements can be useful or misleading.
Nevertheless, many of these assays still do not take into account the solid state form to which the measured solubility refers. The solid state form can be characterized using a variety of analytical techniques, such as powder X-ray diffraction (PXRD), differential scanning calorimetry (DSC), polarized light microscopy or a number of spectroscopic techniques [15]. Before discussing the choice of the most appropriate analytical technique, attention must be paid to the sample to be investigated, the most important information regarding the solid state form and the requirements of the analytical technique. As regards the information obtained from assessment of the solid state form, the distinction between crystalline and amorphous phases is most crucial as this will largely govern thermodynamic solubility. As discussed above, amorphous and crystalline phases can give solubilities, which are orders of magnitude different, whereas this effect is much smaller for different polymorphic or pseudopolymorphic forms. The solid state form of the compound as used for determination of the solubility or the solid state form of the residue obtained from phase separation could be investigated. Figure 2 summarizes scenarios that may be encountered during incubation of a research compound in an aqueous buffer system. The compound as introduced into the buffer system may be either crystalline or amorphous. If it is crystalline, the material will remain in the crystalline state during the incubation since conversion to an amorphous form is very unlikely as this would be accompanied by an increase in free enthalpy. If the research compound is introduced as an amorphous material, it could either remain in an amorphous state or crystallize during the incubation. In the latter case, the measured thermodynamic solubility will undoubtedly refer to the crystalline state and it can be considered as meaningful. However, if the compound remains in an amorphous state, the result obtained must be treated carefully as it does not refer to a crystalline phase, which is desirable for clinical development.
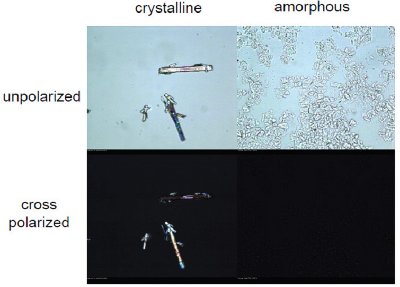
Figure 3 - Assessment of crystallinity by polarized light microscopy. Left hand side: Crystalline residue from thermodynamic solubility assay. Right hand side: Amorphous residue from thermodynamic solubility assay
As regards the analytical techniques used for obtaining information on the solid state form, PXRD might at first glance be considered the most appropriate. The advantage of this method is clearly the easy differentiation between amorphous and crystalline phases as well as different crystalline phases. However – leaving aside the high cost of the method – it must be considered that such in-depth information on the respective crystalline phase might not be particularly useful. During compound optimization, a large number of compounds are typically investigated by measurement of the thermodynamic solubility, and there is very limited knowledge on the different crystalline phases of these compounds. The thermodynamically stable crystal form is generally not known at this stage. Moreover, the demand of PXRD in terms of amount of compound is even today still in the range of several milligrams. As the thermodynamic solubility can be determined using about two milligrams and the solid residue obtained after the phase separation step is of course less than that amount, use of PXRD for distinction between amorphous and crystalline material would increase the amount of compound required for the assay. The same holds true for thermoanalytical techniques such as DSC. Spectroscopic techniques, as infrared, near-infrared or Raman spectroscopy, also have the drawback that it might be not straightforward to distinguish between crystalline and amorphous phases by generic methods. Nevertheless, the old fashioned technique of polarized light microscopy could be very useful in this situation. It clearly has a number of advantages, such as allowing easy distinction between crystalline and amorphous material, as shown in Figure 3, the ability to manage with tiny amounts of solid residue in the microgram range, and very high efficiency in terms of workload.
Accordingly, assays used for determination of thermodynamic solubility can deliver more in-depth information if they are combined with assessment of the solid state form of the residue. Polarized light microscopy is therefore considered to be a valuable technique which is efficient in terms of consumption of compound, workload, throughput and low investment in equipment. It represents a useful extension of thermodynamic solubility assays, which ensures optimization of solubility based on crystalline compounds.
Summary
Optimization of the solubility of research compounds might at first glance appear to rely on very simple methods. However, to ensure that the optimization of the solubility of research compounds goes in the right direction, it is necessary to understand clearly the correlation between solid state properties and their influence on measured solubility. This includes very basic considerations, such as differentiation between measurements of kinetic and thermodynamic solubility, which are clearly intended for different purposes – “does it precipitate?” or “does it dissolve?” – but also includes further assessment of solid state forms in assays for determination of the thermodynamic solubility. There is therefore no need for expensive navigation systems – old-fashioned signposts, such as polarized light microscopy, can do a good job and make sure that one stays on track.
References
- H. van de Waterbeemd, H. Lennernäs, P. Artursson, “Drug Bioavailability”; Wiley-VCH: Weinheim, 2004.
- W. Curatolo, “Physical chemical properties of oral drug candidates in the discovery and exploratory development settings”, Pharm. Sci. Tech. Today 1998, 1, 387-393.
- A. Avdeef, “Physicochemical profiling – solubility, permeability and charge state”, Curr. Top. Med. Chem. 2001, 1, 277-351.
- E.H. Kerns, L. Di, “Drug-like Properties: Concepts, Structure Design and Methods“, Academic Press, 2008.
- C.J. Lipinski, “Drug-like properties and the causes of poor solubility and poor permeability”, Pharmacol. Toxicol. Methods 2000, 44, 235-249.
- E.H. Kerns, “High-throughput physicochemical profiling for drug discovery”, J. Pharm. Sci. 2001, 90, 1838-1858.
- B. Faller, L. Urban, ”Hit and Lead Profiling“; Wiley-VCH: Weinheim, 2009.
- R. Mannhold, “Molecular Drug Properties“; Wiley-VCH: Weinheim, 2008.
- B. Hoelke, S. Gieringer, M. Arlt, C. Saal, ”Comparison of Nephelometric, UV-Spectroscopic, and HPLC Methods for High-Throughput Determination of Aqueous Drug Solubility in Microtiter Plates”, Anal. Chem. 2009, 81, 3165-3172.
- C.D. Bevan, R.S. Lloyd, “A High-Throughput Screening Method for the Determination of Aqueous Drug Solubility Using Laser Nephelometry in Microtiter Plates”, Anal. Chem. 2000, 72, 1781-1787.
- M. Pudipeddi, A.T.M. Serajuddin, “Trends in Solubility of Polymorphs”, J. Pharm. Sci. 2005, 94, 929-939.
- J. Bauer, S. Spanton, R. Henry, J. Quick, W. Dziki, W. Porter, J. Morris, ”Ritonavir: An Extraordinary Example of Conformational Polymorphism“, Pharm. Res. 2001, 18, 859-866.
- R.E. Dinnebier, P. Sieger, H. Nar, K. Shankland, W.I.F. David, ”Structural Characterization of Three Crystalline Modifications of Telmisartan by Single-Crystal and High-Resolution X-ray Powder Diffraction”, J. Pharm. Sci. 2000, 89, 1465-1579.
- A. Glomme, J. Maerz, J.B. Dressman, ”Comparison of a Miniaturized Shake-Flask Solubility Method with Automated Potentiometric Acid/Base Titrations and Calculated Solubility”, J. Pharm. Sci. 2005, 94, 1-16.
- R. Hilfiker, “Polymorphism in the Pharmaceutical Industry”, Wiley-VCH, Weinheim, 2006.
Christoph Saal studied chemistry at the Technical University of Darmstadt with a Ph.D. in Physical Chemistry. Currently he holds a position as Group Leader Molecule Characterization at Merck KGaA. Christoph is a member of EDQM expert groups for spectroscopic methods and holds a teaching position in Pharmaceutical Technology at the Goethe University Frankfurt. Readers may contact the author directly at: [email protected]
This article was printed in the May/June 2010 issue of American Pharmaceutical Review - Volume 13, Issue 4. Copyright rests with the publisher. For more information about American Pharmaceutical Review and to read similar articles, visit www.americanpharmaceuticalreview.com and subscribe for free.