David J. Brayden- UCD Conway Institute, Veterinary Sciences Centre, University College Dublin, Belfield, Dublin, Ireland
Rationale
Oral administration of peptides for systemic delivery offers convenience for patients and promotes better adherence to dosing regimens than injections. While this may seem obvious in principle, the preference of Type II diabetic patients for a once-daily oral tablet over a once-weekly sub-cutaneous (SC) injection of the anti-diabetic Glucagon-like peptide 1 (GLP-1) analogue, semaglutide (Ozempic®), may not be quite so clear-cut when the dosing issues around food interactions are considered.1 In the semaglutide example, patients have two options for administration so the overall uptake of semaglutide by patients will therefore be increased. For other molecules, the conversion advantage in switching from a monthly deep muscle low gauge injection of octreotide (Sandostatin® LAR Depot) to twice-daily capsules (Mycappsa®) is very much in favor of the latter as a preference.2 In treating Type II diabetes, patient psychological aversion to the lifestyle change to daily insulin and GLP-1 injections can delay take up of these highly efficacious molecules. It is estimated that a considerable percentage of the public are needle phobic.3 With an oral version of a GLP-1 analogues being available, Type II diabetic patients will likely move to such options earlier in their disease and beneficial outcomes will be improved compared to if they delay take-up due to the psychological barrier of injections. Commercially, oral versions of injectable peptides also allow new IP to be generated on new formulations and offer competitive advantages.
Peptides and proteins are large molecules with high potency and high specificity for their drug targets with a tendency for low off-target side-effects compared to small molecules. They are expensive to synthesise by the processes of solid phase synthesis or recombinant expression, the manufacturing method being dependent on chain length. Recombinant expression is in turn cheaper than synthetic processes. The vast majority of approved peptides are administered by injection,4 many in concentrations of 1 mg or less per week. Assuming oral bioavailability of ~1% with current formulations that have been FDA-approved compared to injections of the same peptide, the cost-of-goods argument becomes unsustainable for most candidates that might be considered for conversion to the oral route. For the approved semaglutide products, there is a ~100-fold increase in weekly dose requirements when switching from the typical dose of SC administration (1 mg) to oral (98 mg),5 reflecting a considerable manufacturing cost challenge for the latter. Assuming high potency as the starting point, very low and variable oral bioavailability are therefore the main reasons that there are so few current FDA-approved oral formulations of peptides for systemic delivery (Table 1).
GI Physiology
Linear peptides can be attacked by acid and pepsin in the stomach. If they survive gastric transit, they have to negotiate pancreatic enzymes in the duodenum. They then encounter peptidases in the brush border of the upper GI epithelium, along with enterocyte intracellular enzymes. In digesting protein, the GI serves them up to small intestinal epithelia mainly as di- and tripeptide substrates for the PEPT1- and other enterocyte carriers. Replacement of L-amino acids with D-amino acids, methylation of the N-terminus, and use of cyclic residues can improve peptide stability to an extent, along with formulation approaches including enteric coating and inclusion of excipients that change local pH away from the enzyme peptidase activity.6 Peptides and proteins have large molecular weights (MW) and tend to be highly polar, features that mitigate against intestinal transepithelial flux. Some low MW hydrophilic peptides can permeate epithelial tight junctions (TJ), however basal paracellular flux is of low capacity. Reversibly opening TJs with enhancers remains a strategy actively being pursued,but for the most part enhancers that have reached clinical trials as part of oral peptide formulations seem to have multimodal effects on the epithelium initiated by induction of plasma membrane fluidization.7 Similarly, hydrophobic stable peptides designed with lipidic fatty acid appendages to be long-acting should dissolve better in lipid bilayers, but they do not seem to permeate sufficiently in the absence of enhancers to achieve adequate blood levels in vivo.8 Finally, peptide structures designed with affinity for PEPT1 and other carriers have not yet achieved the same bioavailability of similarly designed small molecules. In sum, the successful achievement of oral bioavailability for peptides must overcome the pH challenges of the stomach, a host of attacking peptidases in the GI lumen and brush border, low epithelial permeability, as well as the liver first pass effect (Figure 1). The unusual macrocyclic peptides, cyclosporine and voclosporin detailed in Table 1, have much higher oral bioavailability than typical linear peptides. They are inherently GI-stable and lipophilic; their main drawback is metabolism by cytochrome P450 and efflux by P-glycoprotein in the gut wall, rather than having low permeability.9

The excipients in oral cyclosporin Neoral® (Novartis, Basel, Switzerland) were therefore designed primarily to improve solubility and inhibit GI metabolism and efflux, not permeability.
Suitable Peptide Candidates
Since oral bioavailability (F) is the product of the drug fraction absorbed, the liver first pass effect and liver blood flow, we can work out the maximum achievable value for F.
Fmax achievable = Fa (absorbed). Fg (gut). Fh (liver)
Fh is what survives the first pass effect (Eh ), hence…
Fh = 1 –Eh
Eh = CL / Qh (CL: hepatic clearance; Q: hepatic blood flow)
So, plugging in: Fmax = Fa. (1 – (CL / Qh))……(assuming Fg =1). Adapted from Reference 10.
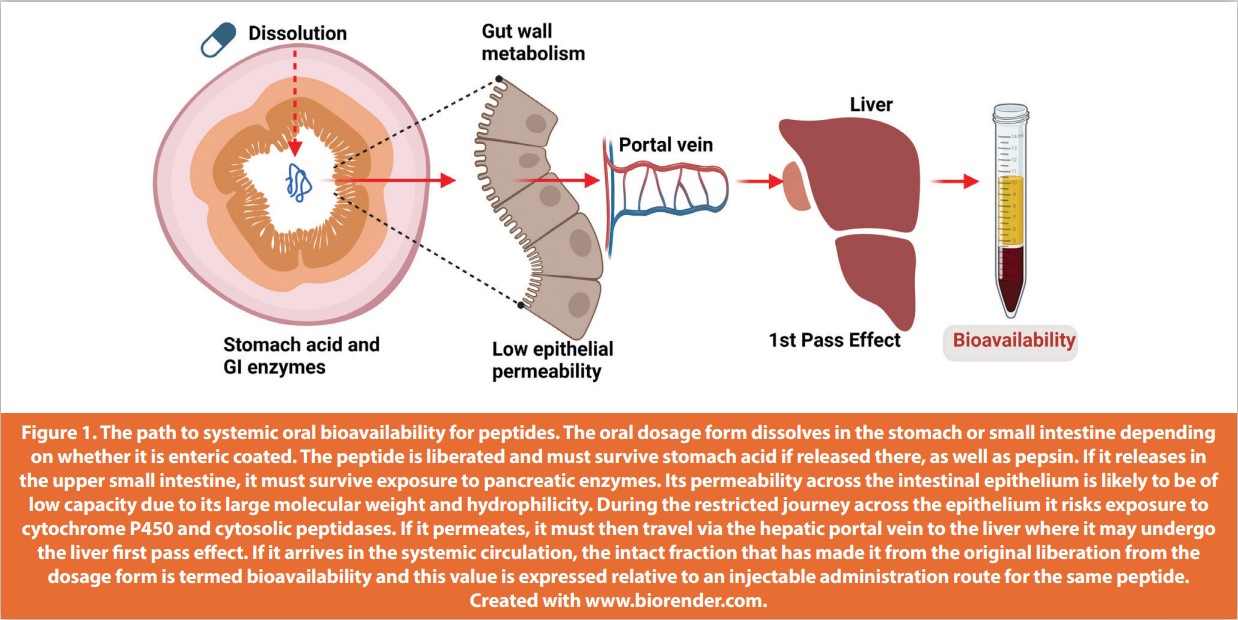
If this Fmax value is too low, no delivery technology will suffice. For peptide candidates that pass that calculation step, extremely high potency is essential as otherwise too high a blood level would be required for efficacy and this will make formulation impractical. Octreotide (MW 1019 Da) offered the advantage of a relatively small increase in stability in intestinal fluid afforded by a macrocycle backbone, two D-amino acids, and a modified C terminus,11 whereas semaglutide remained unstable in the GI tract despite being stable in plasma due to affinity of its fatty acid chains for albumin. The key to why semaglutide was such a good candidate for oral formulation lies in its long plasma half-life (t½) of 165 hours. This means that an oral bioavailability of 1% can suffice as a top-up once steady state plasma levels are achieved in 4-5 weeks; missing a dose or a failure to deliver from a dose would have no material impact on blood levels.12 Finally, another consideration is that some peptides require pulsatile delivery to plasma rather than sustained levels; this applies to parathyroid hormone (PTH) (1-34). Often suggested as a candidate for oral administration to treat osteoporosis,13 this can be a confounding factor that adds to the hurdles for formulating PTH (1-34) in an oral dosage form. In sum, ideal peptide candidates for oral administration should be at the lower end of the MW spectrum for large molecules, be potent, have some inbuilt GI stability, have long t½ values, and be synthesised at reasonable cost. Apart from the atypical peptides, cyclosporine and voclosporin, no other formulated candidate produced to date has all of these features.
Permeation Enhancers
Table 2 provides a summary of other peptides and associated oral administration formulation strategies that have advanced to clinical trials. They are dominated by permeation enhancers, assisted in many cases by enteric coatings and the presence of peptidase-inhibiting excipients or direct-acting inhibitors. Medium chain fatty acids (sodium caprylate C8 , sodium caprate, C10) and derivatives (the Eligen® candidate, salcaprozate sodium (SNAC)) along with EDTA, bile salts, and acyl carnitines are the main examples of enhancers. For oral semaglutide (Rybelsus®, Novo Nordisk, Denmark), SNAC seems to provide a major benefit from inhibiting pepsin in the stomach, along with a degree of transcellular permeability enhancement across gastric parietal cells.14 No enteric coating is required when using SNAC, and it can also act as a small intestinal enhancer with other payloads.
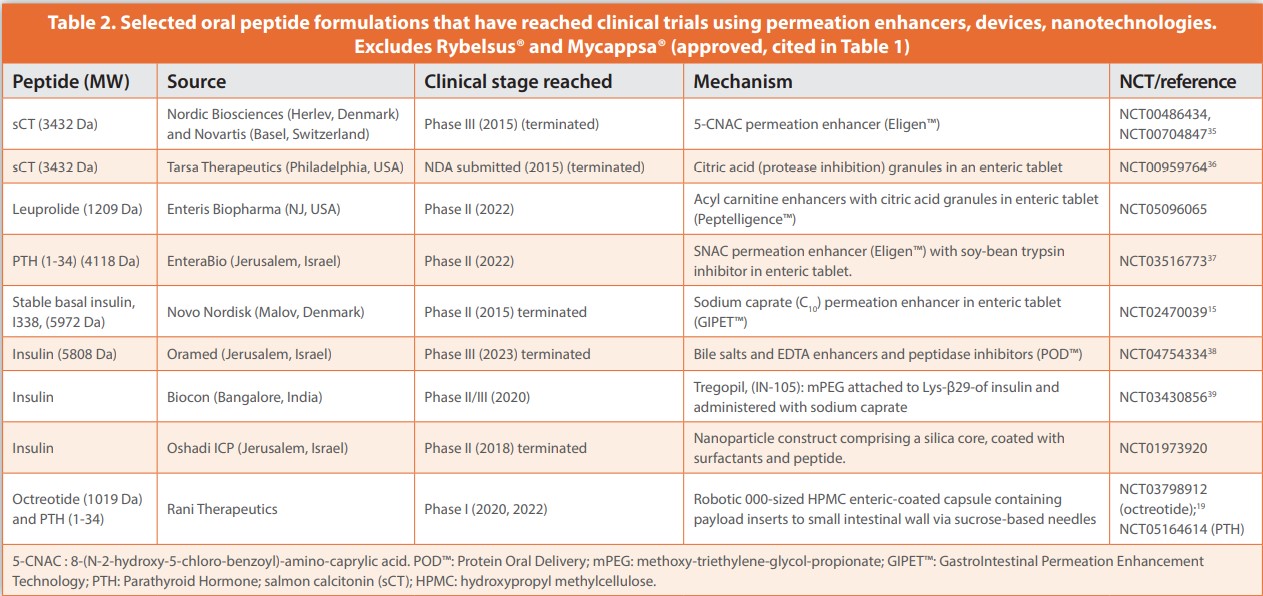
C8 is a key component of the Transient Permeation Enhancer™ (TPE™) system of Chiasma (Israel), an emulsion-based system that enables the oral octreotide formulation.11 TPE™ seems to act mainly by opening tight junctions according to studies using Caco-2 monolayers and a cannulated rat model. C10 has been formulated in enteric tablets with a range of peptides and antisense oligonucleotides, however to date, despite average F values that have exceeded those of SNAC and C8 in some cases,15,16 it has yet to be marketed in an approved oral product. C10 has a multimodal action in vivo, initiated by perturbation of intestinal epithelial plasma membranes by micellar constructs leading to contraction of the apical actin ring at TJs.17 These traditional enhancers provide a maximum low single digit F with high variability for peptides, which will not satisfy requirements for most candidates. The industry is caught in a bind as it defaults to using enhancers with excipient status or a history of use in humans rather than experimenting with new ones with unknown toxicology and higher regulatory costs and risks. Perhaps the FDA’s pilot PRIME initiative on a pathway to approving excipients independent of associated bioactive molecules will provide new options for enhancer selection.18 To date, clinical trials for peptides formulated with enhancers have not provided evidence of co-absorption of pathogens as once feared, likely due to the transient nature of the mechanism and because opened TJs have a MW- and size capacity for fluxes. More safety aspects should be revealed following chronic administration in approved peptide products.
Devices
Research on devices for oral peptide delivery was a reaction to the apparent ceiling in maximum F values that could be achieved with enhancers. Leveraging from the transdermal microneedle field, an orally-administered robotic pill was developed by Rani Therapeutics (USA) in which sucrose-based needles were activated from a capsule following an osmotic gas-driven swelling. This led to a Phase 1 trial for octreotide where over 60% F was achieved from those pills that actuated in the small intestine (Table 1).19 MIT and Novo Nordisk designed a stomach-targeted device (SOMA),20 which can orient itself and whose needle was also was actuated by fluid. The latest version, L-SOMA, has delivered elevated levels of insulin, protein, and antibodies in an endoscopic-assisted pig model.21 Other examples include a hooking device for the stomach from Biograil (Denmark) and devices using ultrasound (Baywind Bio, USA). There is no doubt that the delivery achieved from such devices far exceeds that of permeation enhancer-based tablets and capsule formulations. Yet, only the Rani system has reached clinical trials to date. Many hurdles for approval of devices remain including low mg loadings, potential to block the lumen of the GI tract with 000 size capsules, the possibility of gut wall perforation, an unclear regulatory pathway, and the question of whether patients will ultimately find these approaches acceptable.
Nanotechnology
Nanotechnology has undergone a cyclical history in oral peptide administration. Currently, optimism is at a low point. Just a few prototypes ever reached clinical trials including a liposome-containing insulin targeted to liver cells, a silica-based particle with insulin, and a calcium phosphate-based system.22 However, none of these systems appear to still be in clinical development. Despite much excitement in an FcRn receptor-targeted nanoparticle for insulin,23 there is little sign of widespread activity. A bile salt-coated nanoparticle is in development, with encouraging data seen for insulin in rodents.24 My own group is pursuing a core shell structure with a peptide in a core with L-arginine and zinc, then overcoated with silica.25 Perhaps the best recent preclinical data in rodents has been reported for a micellar nanoparticle with a carboxy-betaine side chain.26 For most of these nanotechnologies, there is little evidence of scale-up and manufacturing potential, while evidence of efficacy in large animal models is scant. Adequate peptide loading of the particles and release at the right time and place are issues. For GI-receptor targeted prototypes, it is not clear how such a particle avoids surface premature binding from protein and lipids in the GI tract. Moreover, evidence of significant particle enterocyte endocytosis across the gut wall in vivo is scant,27 even if negotiation though the mucus barrier through surface coatings has been addressed to some extent.28 It seems that particles with contrasting characteristics are needed to surmount both these barriers. Such complexity in particle design mitigates the potential for development compared to the more straightforward enhancer-based formulations that they are competing against.
Conclusion
Traditional tablet and capsule formulations of peptides with permeation enhancers have generated the first group of FDA-approved oral peptides for systemic delivery. These types of systems may also suffice for dual GLP-1/GIP agonists if an F of 1% is deemed acceptable.17 Yet, they are very costly in terms of peptide waste. Oral semaglutide and octreotide, if further developed as second-generation products, will likely require reductions of peptide dose in more efficiently-delivered formulations in the long term. This might be achieved with newly discovered enhancers or combinations of existing ones with different mechanisms of action. Looking beyond GLP-1 and insulin as payloads (a molecule that increasingly appears to be a poor commercial candidate),29 at least ten companies are working on device-based solutions to administer proteins and monoclonal antibodies orally and some technologies including the RaniPill™ have reached Phase I.19 While these offer more than a ten-fold increase in F over permeation enhancer-based systems, they have their own issues in terms of reliability of activation at the gut wall, safety, and an uncertain regulatory pathway. Nanotechnology has even higher hurdles to overcome including complex syntheses with expensive reagents some of which will not be GRAS, as well as lack of quantitative evidence of particle uptake in vivo, but at least the GI physiology aspects are now better understood along with improved in vivo imaging. These three approaches will continue to be assessed in the context of the potent and stable peptide candidates with lower molecular weights that are emerging from medicinal chemistry than have been available before.
Acknowledgment and Declaration
The author acknowledges funding support from the Science Foundation Ireland (SFI) Centre for Medical Devices (CURAM) (Grant no. 13/RC/2073_P2) and by the European Union Regional Development Fund, and also from Enterprise Ireland Commercialisation Fund, grant number CF-2020-1556.
References
- Brunton SA, Mosenzon O, Wright EE Jr. Integrating oral semaglutide into clinical practice in primary care: for whom, when, and how? Postgrad. Med. 2020: 132(suppl.2):48-60. doi: 10.1080/00325481.2020.1798162.
- Yuen KCJ, Samson SL. Oral octreotide: a review of recent clinical trials and practical recommendations for its use in the treatment of patients with acromegaly. Endocr. Pract. 2022: 28(6):637-645. doi: 10.1016/j.eprac.2022.04.009.
- Alsbrooks K, Hoerauf K. Prevalence, causes, impacts, and management of needle phobia: An international survey of a general adult population. PLoS One. 2022: 17(11):e0276814. doi: 10.1371/journal.pone.0276814.
- Muttenthaler M, King GF, Adams DJ, Alewood PF. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021: 20(4):309-325. doi: 10.1038/s41573-020-00135-8.
- Aroda VR, Blonde L, Pratley RE. A new era for oral peptides: SNAC and the development of oral semaglutide for the treatment of type 2 diabetes. Rev. Endocr. Metab. Disord. 2022: 23(5):979-994. doi: 10.1007/s11154-022-09735-8.
- Brayden DJ, Hill TA, Fairlie DP, Maher S, Mrsny RJ. Systemic delivery of peptides by the oral route: Formulation and medicinal chemistry approaches. Adv. Drug Deliv. Rev. 2020;157:2- 36. doi: 10.1016/j.addr.2020.05.007.
- Maher S, Brayden DJ. Formulation strategies to improve the efficacy of intestinal permeation enhancers. Adv. Drug Deliv. Rev. 2021:177:113925. doi: 10.1016/j.addr.2021.113925.
- Kjeldsen TB, Hubálek F, Tagmose TM, et al. Engineering of orally available, ultralong-acting insulin analogues: discovery of OI338 and OI320. J. Med. Chem. 2021: 64(1):616-628. doi: 10.1021/acs.jmedchem.0c01576.
- Benet LZ. The drug transporter-metabolism alliance: uncovering and defining the interplay. Mol. Pharm. 2009: 6(6):1631-43. doi: 10.1021/mp900253n.
- Rautio J, Kumpulainen H, Heimbach T, et al. Prodrugs: design and clinical applications. Nat. Rev. Drug Discov. 2008: 7(3):255-70. doi: 10.1038/nrd2468.
- Brayden DJ, Maher S. Transient Permeation Enhancer® (TPE®) technology for oral delivery of octreotide: a technological evaluation. Expert Opin. Drug Deliv. 2021: 18(10):1501- 1512. doi: 10.1080/17425247.2021.1942838.
- Berg S, Edlund H, R F Goundry W, et al. Considerations in the developability of peptides for oral administration when formulated together with transient permeation enhancers. Int. J. Pharm. 2022: 628:122238. doi: 10.1016/j.ijpharm.2022.122238.
- Sturmer A, Mehta N, Giacchi J, et al. Pharmacokinetics of oral recombinant human parathyroid hormone [rhPTH(1-31)NH₂] in postmenopausal women with osteoporosis. Clin. Pharmacokinet. 2013: 52(11):995-1004. doi: 10.1007/s40262-013-0083-4.
- Buckley ST, Bækdal TA, Vegge A, et al. Transcellular stomach absorption of a derivatized glucagon-like peptide-1 receptor agonist. Sci. Transl. Med. 2018: 10(467):eaar7047. doi: 10.1126/scitranslmed.aar7047.
- Halberg IB, Lyby K, Wassermann K, et al. Efficacy and safety of oral basal insulin versus subcutaneous insulin glargine in type 2 diabetes: a randomised, double-blind, phase 2 trial. Lancet Diabetes Endocrinol. 2019: 7(3):179-188. doi: 10.1016/S2213-8587(18)30372-3.
- Raoof AA, Chiu P, Ramtoola Z, et al. Oral bioavailability and multiple dose tolerability of an antisense oligonucleotide tablet formulated with sodium caprate. J. Pharm. Sci. 2004: 93(6):1431-9. doi: 10.1002/jps.20051.
- Tran H, Aihara E, Mohammed FA, et al. In Vivo mechanism of action of sodium caprate for improving the intestinal absorption of a GLP1/GIP co-agonist peptide. Mol. Pharm. 2023 Jan 2. doi: 10.1021/acs.molpharmaceut.2c00443.
- https://www.fda.gov/drugs/development-approval-process-drugs/pilot-program[1]review-innovation-and-modernization-excipients-prime Accessed January 27th, 2023.
- Dhalla AK, Al-Shamsie Z, Beraki S, et al. A robotic pill for oral delivery of biotherapeutics: safety, tolerability, and performance in healthy subjects. Drug Deliv. Transl. Res. 2022; 12(1):294-305. doi: 10.1007/s13346-021-00938-1.
- Abramson A, Caffarel-Salvador E, Khang M, et al. An ingestible self-orienting system for oral delivery of macromolecules. Science. 2019 : 363(6427):611-615. doi: 10.1126/science. aau2277
- Abramson A, Frederiksen MR, Vegge A. et al. Oral delivery of systemic monoclonal antibodies, peptides and small molecules using gastric auto-injectors. Nat. Biotechnol. 2022: 40(1):103-109. doi: 10.1038/s41587-021-01024-0.
- Aguirre TA, Teijeiro-Osorio D, Rosa M, et al. Current status of selected oral peptide technologies in advanced preclinical development and in clinical trials. Aguirre TA, Teijeiro-Osorio D, Rosa M, et al. Adv. Drug Deliv. Rev. 2016: 106(Pt B):223-241. doi: 10.1016/j. addr.2016.02.004.
- Pridgen EM, Alexis F, Kuo TT, et al. Transepithelial transport of Fc-targeted nanoparticles by the neonatal fc receptor for oral delivery. Sci. Transl. Med. 2013: 5(213):213ra167. doi: 10.1126/scitranslmed.3007049.
- Lee JS, Han P, Chaudhury R, Khan S, et al. Metabolic and immunomodulatory control of type 1 diabetes via orally delivered bile-acid-polymer nanocarriers of insulin or rapamycin. Nat. Biomed. Eng. 2021: 5(9):983-997. doi: 10.1038/s41551-021-00791-0.
- Hristov D, McCartney F, Beirne J, et al. Silica-coated nanoparticles with a core of zinc, l-arginine, and a peptide designed for oral delivery. ACS Appl. Mater. Interfaces. 2020 : 12(1) :1257-1269. Doi : 10.1021/acsami.9b16104.
- Han X, Lu Y, Xie J, et al. Zwitterionic micelles efficiently deliver oral insulin without opening tight junctions. Nat. Nanotechnol. 2020: 15(7):605-614. doi: 10.1038/s41565-020-0693-6.
- Lundquist P, Khodus G, Niu Z, et al. Barriers to the intestinal absorption of four insulin[1]loaded arginine-rich nanoparticles in human and rat. ACS Nano. 2022: 16(9):14210-14229. doi: 10.1021/acsnano.2c04330.
- Knoll P, Hörmann N, Nguyen Le NM, et al. Charge converting nanostructured lipid carriers containing a cell-penetrating peptide for enhanced cellular uptake. J. Colloid Interface Sci. 2022: 628(Pt A):463-475. doi: 10.1016/j.jcis.2022.07.160.
- https://oramed.com/oramed-announces-top-line-results-from-phase-3-trial-of-ormd[1]0801-for-the-treatment-of-type-2-diabetes/ Accessed January 27th, 2023.
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/021795s006lbl.pdf Accessed January 30th, 2023.
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/050715s027,050716s028l bl.pdf Accessed January 30th, 2023.
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/213051s000lbl.pdf Accessed January 30th, 2023.
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/208232s000lbl.pdf Accessed January 30th, 2023.
- https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/213716Orig1s000lbl.pdf Accessed January 30th, 2023.
- Karsdal MA, Byrjalsen I, Alexandersen P, et al. Treatment of symptomatic knee osteoarthritis with oral salmon calcitonin: results from two phase 3 trials. Osteoarthritis Cartilage. 2015: 23(4):532-43. doi: 10.1016/j.joca.2014.12.019.
- Binkley N, Bolognese M, Sidorowicz-Bialynicka A, et al. A phase 3 trial of the efficacy and safety of oral recombinant calcitonin: the Oral Calcitonin in Postmenopausal Osteoporosis (ORACAL) trial. J Bone Miner Res. 2012: 27(8):1821-9. doi: 10.1002/jbmr.1602.
- Burshtein G, Itin C, Tang JCY, et al. The combined effect of permeation enhancement and proteolysis inhibition on the systemic exposure of orally administrated peptides: Salcaprozate sodium, soybean trypsin inhibitor, and teriparatide study in pigs. Int J Pharm X. 2021: (3) 100097. doi: 10.1016/j.ijpx.2021.100097.
- Eldor R, Francis BH, Fleming A, et al. Oral insulin (ORMD-0801) in type 2 diabetes mellitus: A dose-finding 12-week randomized placebo-controlled study. Diabetes Obes Metab. 2022 : 1-10. Doi : 10.1111/dom.14901.
- Lebovitz HE, Fleming A, Cherrington AD, et al. Expert Opin Pharmacother. Efficacy and safety of Tregopil, a novel, ultra-rapid acting oral prandial insulin analog, as part of a basal-bolus regimen in type 2 diabetes: a randomized, active-controlled Phase 2/3 study. Expert Opin. Pharmacother. 2022: 23(16):1855-1863. doi: 10.1080/14656566.2022.2141569.
Author Biography
David Brayden is Professor of Advanced Drug Delivery at University College Dublin. His research is in non-injected peptide administration. David spent 10 years as a senior scientist at Elan Biotechnology Research before joining academia. He is a Fellow of the American Association of Pharmaceutical Scientists and the Controlled Release Society. In 2021, David was appointed Chief Editor of Frontiers in Drug Delivery and he is a current member of Ireland’s National Research Ethics Committee (Clinical Trials).
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special
offers from American Pharmaceutical Review delivered to your inbox!
Sign up now!